



Los estudios en el laboratorio han permitido identificar cambios del metabolismo de halógenos en suero y líquido cefalorraquídeo (LCR) de pacientes con enfermedad de Parkinson, que indican la presencia de «autohalogenación acelerada» del LCR de los pacientes o aumento de haloperoxidasas, en concreto, tiroperoxidasa en sangre y lactoperoxidasa en LCR. Además, se ha detectado un exceso en suero y LCR de algunos derivados halogenados, como proteínas con halogenación avanzada tipo advanced oxidation protein products (AOPP). Estos hechos, «autohalogenación acelerada» e incremento de haloperoxidasas y proteínas AOPP, indican la presencia de estrés halogenativo en la enfermedad de Parkinson. Además, un derivado halogenado, la 3-yodo-L-tirosina, muestra toxicidad parkinsoniana en modelos experimentales, pues se ha observado que induce agregados de α-sinucleína y daño de las neuronas de dopamina en cerebro e intestino en ratones. La hipótesis que se maneja es que en la enfermedad de Parkinson existe un exceso halogenativo, relacionado con una alteración haloenzimática de síntesis o degradación de oxiácidos de halógenos y sus derivados halogenados. Este estrés halogenativo se relacionaría con el daño del sistema nervioso.
Laboratory studies identified changes in the metabolism of halogens in the serum and cerebrospinal fluid (CSF) of patients with Parkinson's disease, which indicates the presence of «accelerated self-halogenation» of CSF and/or an increase in haloperoxidases, specifically serum thyroperoxidase and CSF lactoperoxidase. Furthermore, an excess of some halogenated derivatives, such as advanced oxygenation protein products (AOPP), has been detected in the CSF and serum. «Accelerated self-halogenation» and increased levels of haloperoxidases and AOPP proteins indicate that halogenative stress is present in Parkinson's disease. In addition, 3-iodo-L-tyrosine, a halogenated derivative, shows «parkinsonian» toxicity in experimental models, since it has been observed to induce α-synuclein aggregation and damage to dopaminergic neurons in the mouse brain and intestine. The hypothesis is that patients with Parkinson's disease display halogenative stress related to a haloenzymatic alteration of the synthesis or degradation of oxyacid of halogens and their halogenated derivatives. This halogenative stress would be related to nervous system damage.
El estrés oxidativo se define como un desequilibrio entre la producción de especies reactivas de oxígeno y los sistemas antioxidantes, y se considera actualmente como un mecanismo patogénico importante en la enfermedad de Parkinson (EP)1,2. Entre los diversos tipos de estrés oxidativo3 destacan el estrés peroxidativo, el de especies reactivas con nitrógeno (como el óxido nítrico o •NO) y el estrés de especies halogenadas (como el ácido hipocloroso o HOCl).
El estrés peroxidativo se debe a exceso de ión superóxido (•O2−) o agua oxigenada (H2O2) y se detecta en tejido cerebral, sangre y líquido cefalorraquídeo (LCR) de los pacientes con EP. Se sabe que la sustancia negra está sometida a intenso estrés peroxidativo, pues muestra un notable incremento de marcadores oxidativos como lípidos peroxidados4, 8-hidroxiguanosina (marcador de estrés oxidativo del ADN)5, proteínas carboniladas6 y productos de lipooxidación avanzada7. Estudios con LCR muestran un importante descenso de la actividad de diversas enzimas antioxidantes relacionadas con peroxidación en los pacientes con EP8.
Respecto al estrés oxidativo por especies reactivas con nitrógeno, el exceso de actividad de •NO induce 2tipos: el estrés nitrativo y el estrés S-nitrosilativo. En la nitración, la oxidación proteica ocurre en residuos de tirosina, mientras que en la S-nitrosilación sucede en residuos de cisteína. El estrés nitrativo se detecta en la EP y afecta a proteínas muy importantes en la enfermedad como la superóxido-dismutasa tipo 1 (MnSOD), la tirosina-hidroxilasa (TH) y la α-sinucleína (αSYN). La MnSOD nitrada se detecta en los cuerpos de Lewy y en el LCR de los pacientes9. Esta proteína nitrada pierde su función y origina, en ratones, vacuolización mitocondrial y peroxidación lipídica, fenómenos que se observan en la EP10. La TH se nitra de modo selectivo en la EP, lo cual inhibe su función enzimática, lo que podría participar en la patogenia de la enfermedad11,12. La αSYN nitrada (3NT-αSYN) es un componente de los cuerpos de Lewy y es una forma anómala de αSYN que se relaciona también con la patogénesis de la EP13,14. Estudios recientes con suero de pacientes con EP indican la presencia de estrés nitrativo, que se pone en evidencia por aumento significativo de proteínas 3-nitrotirosinadas y de 3NT-αSYN15. Finalmente, en la S-nitrosilación, el óxido nítrico se une a sulfidrilos de cisteínas y da lugar a derivados nitroso-sulfidrilos. Esto modifica la funcionalidad de diversas proteínas importantes en la EP, como la proteína disulfuro isomerasa o la parkina16,17. Ambas proteínas nitrosiladas pierden su capacidad antioxidante y de proubiquitinación y se facilita la agregación y depósitos proteicos18,19.
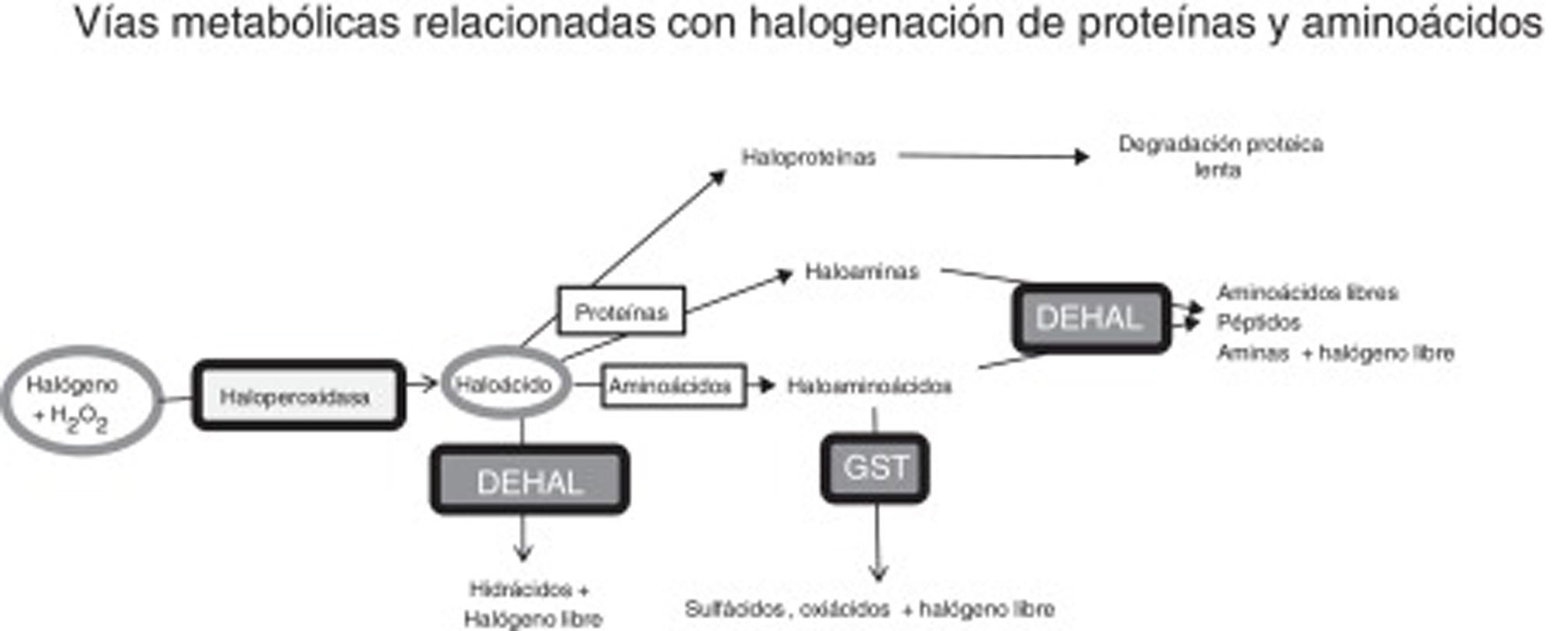
Estrés halogenativo y enfermedad de ParkinsonRespecto al estrés halogenativo, se debe a un exceso de especies reactivas halogenadas, principalmente oxiácidos de halógenos, como ácido hipocloroso o ácido hipoyodoso. Las haloperoxidasas son las enzimas que catalizan la conversión de agua oxigenada en oxiácidos, tras la incorporación de halógenos (fig. 1)20-23. Las especies halogenadas son de creciente importancia para entender la etiología de la EP y otras enfermedades neurodegenerativas1,3,24-26. Entre las haloperoxidasas, destacan las de células blancas y microglía, como la mieloperoxidasa y la eosinófilo-peroxidasa; o las de células captadoras de yodo/bromo como tiroides, glándulas salivares y mama, como la tiroperoxidasa (TPO), peroxidasa salival y la lactoperoxidasa (LPO). Los oxiácidos, a su vez, halogenan aminoácidos y proteínas, de modo que estos derivados se incrementan si hay exceso de oxiácidos o de actividad haloperoxidasa.

Vías metabólicas relacionadas con estrés halogenativo, que afectan a proteínas y aminoácidos. Las haloperoxidasas producen oxiácidos de halógenos a partir de halógenos y agua oxigenada. Los oxiácidos son degradados por deshalogenasas a hidrácidos o pueden, a su vez, halogenar proteínas y aminoácidos. Las proteínas se convierten en haloproteínas o haloaminas, y los aminoácidos en haloaminoácidos. Las haloproteínas se degradan lentamente. Las haloaminas y los haloaminoácidos son degradados por deshalogenasas, generando aminoácidos, péptidos y aminas, y por la glutatión-S-transferasa, generando sulfácidos y oxiácidos. En fin, un exceso de actividad haloperoxidasa o un defecto de degradación enzimática pueden causar estrés halogenativo, con exceso de oxiácidos y derivados halogenados.
DEHAL: deshalogenasas; GST, glutatión-S-transferasa; H2O2: agua oxigenada.
Entre los aminoácidos halogenados, aquellos que se derivan de tirosina como clorotirosinas o yodotirosinas son de sumo interés para la EP, pues han demostrado toxicidad sobre neuronas de dopamina y son inhibidores de la tirosina-hidroxilasa1,25,27-29. Otros aminoácidos clorados, como clorocisteína o clorolisina, también dañan las neuronas de dopamina al alterar las proteínas de membrana30. Dentro de la familia de proteínas halogenadas destacan las proteínas modificadas por hipoclorito, las haloaminas y los productos proteicos con halogenación avanzada o AOPP (en inglés advanced oxidation protein products)31. Las proteínas modificadas por hipoclorito presentan intensa cloración en residuos de cisteínas. Las haloaminas son oligopéptidos halogenados. Las AOPP son proteínas anormales que presentan fuerte halogenación, sobre todo clorada, de residuos ditirosínicos y de lisina de la molécula.
En nuestro laboratorio se ha detectado en suero un incremento de TPO en ∼35% de los pacientes15, y en LCR un incremento de LPO en ∼43% de los pacientes (no publicado). También hay que citar que los pacientes suelen presentar exceso de AOPP en suero y LCR15. Todo ello indica que hay estrés halogenativo en los pacientes con EP. Los oxiácidos de halógenos y sus derivados amínicos (haloaminoácidos y haloaminas) son degradados principalmente por deshalogenasas (DEHAL), aunque también interviene la glutatión-S-transferasa (GST). Entre las deshalogenasas destacan las yodotirosina-deshalogenasas tipo 1 o DEHAL1. Las yodotirosina-deshalogenasas 1 presentan 2isoformas principales (DEHAL 1 y 1B) y son oxidorreductasas que en humanos se encuentran codificadas por el gen IYD32. Los derivados halogenados proteicos, que suelen aparecer en situaciones de estrés halogenativo mantenido como inflamación crónica o desórdenes metabólicos, son degradados lentamente y permanecen en los fluidos31. La figura 1 muestra las vías de metabolismo y estrés halogenativo comentadas.
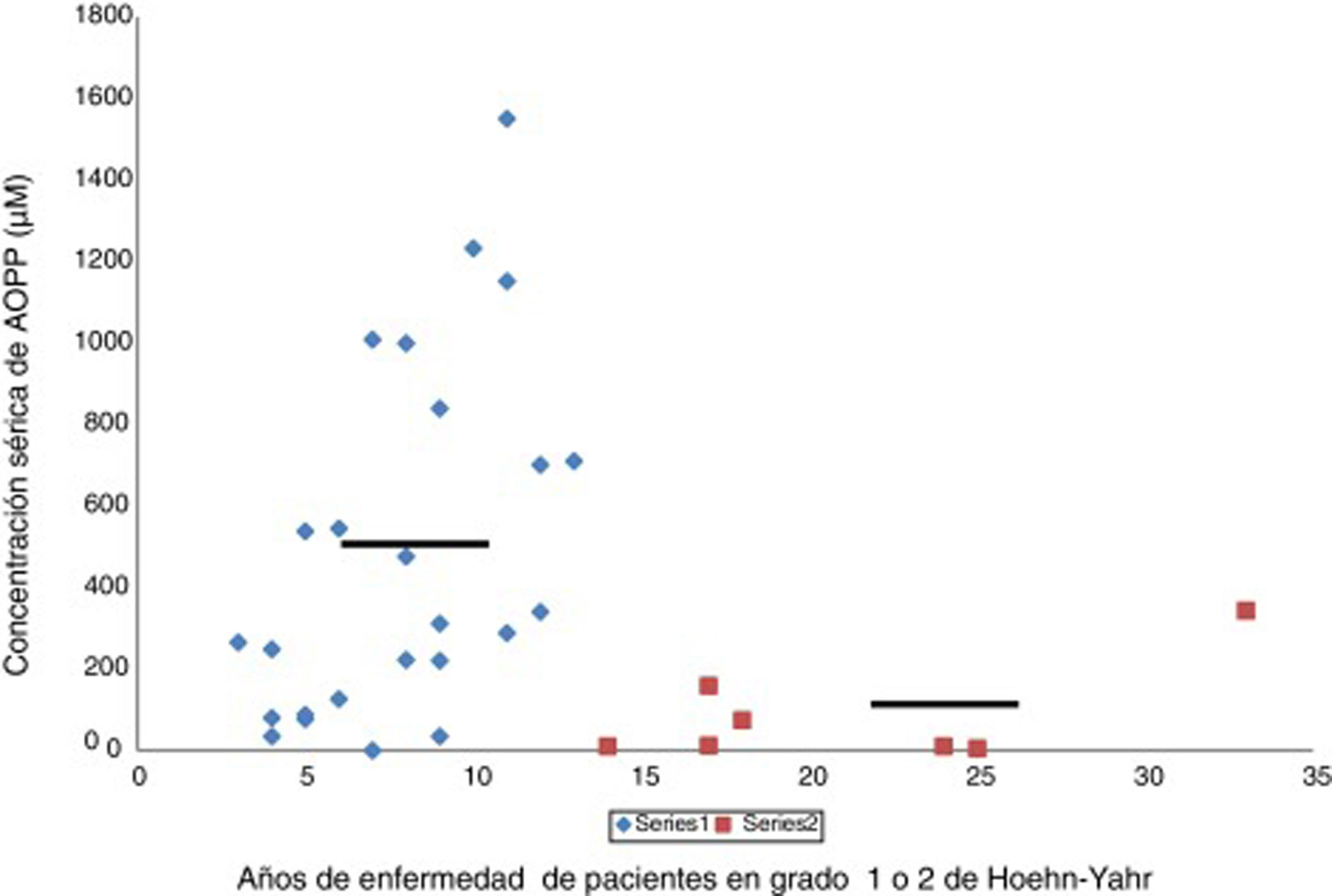
En la enfermedad de Parkinson hay una actividad halogenativa anómalaLos estudios en el laboratorio han permitido identificar cambios halogenativos en suero y LCR de los pacientes. Primeramente se detectó en suero un exceso de proteínas con halogenación avanzada tipo AOPP. Además, los niveles de AOPP séricos se relacionan con el tiempo que la enfermedad permanece en grado 2 de Hoehn-Yahr sin pasar a estadios más avanzados, como muestra la figura 2. Los enfermos con más de 13años con EP y que se encuentren en grado 2, o sea, en un relativo buen estado motor, presentan de modo significativo bajos niveles séricos de AOPP. Estos pacientes podrían poseer mecanismos antihalogenativos más eficaces o un estrés halogenativo global menos severo. En este sentido, hay que añadir que no se detectaron cambios de interés en la mieloperoxidasa sérica o del LCR, la haloperoxidasa más relacionada con la producción de AOPP25.
 se encuentran en la etapa de enfermedad de Parkinson grado 1 o 2 de Hoehn-Yahr. Se observa que aquellos pacientes con una duración de la enfermedad de más de 13 años presentan bajos niveles séricos de AOPP (rombos, pacientes < 13 años, AOPP 487 ± 60μM; cuadrados, pacientes con más de 13 años de evolución, AOPP 87 ± 9μM; p < 0,01, test de Student). Las líneas continuas representan la media de cada grupo. La escala de Hoehn-Yahr es un sistema comúnmente utilizado para describir cómo progresan los síntomas de la enfermedad de Parkinson (desde 1, afectación unilateral solamente, hasta 5, confinamiento en la cama o silla de ruedas a menos que lo ayuden). Fuente: Modificado García-Moreno et al.25")
Relación entre los niveles séricos de proteínas con halogenación avanzada tipo AOPP y la duración correspondiente de la enfermedad de Parkinson en años. Los pacientes (n = 34) se encuentran en la etapa de enfermedad de Parkinson grado 1 o 2 de Hoehn-Yahr. Se observa que aquellos pacientes con una duración de la enfermedad de más de 13 años presentan bajos niveles séricos de AOPP (rombos, pacientes < 13 años, AOPP 487 ± 60μM; cuadrados, pacientes con más de 13 años de evolución, AOPP 87 ± 9μM; p < 0,01, test de Student). Las líneas continuas representan la media de cada grupo. La escala de Hoehn-Yahr es un sistema comúnmente utilizado para describir cómo progresan los síntomas de la enfermedad de Parkinson (desde 1, afectación unilateral solamente, hasta 5, confinamiento en la cama o silla de ruedas a menos que lo ayuden).
Fuente: Modificado García-Moreno et al.25
Además, se observó la presencia de AOPP en LCR de los enfermos, cuando era indetectable en LCR de sujetos control. Se detectó AOPP en LCR de ∼53% de los pacientes, con un contenido medio de AOPP de 11,4 ± 2μM. No se observó correlación con el grado de la enfermedad ni con los años de evolución, pero la presencia de proteínas AOPP en LCR indica, sin duda, estrés halogenativo en el sistema nervioso central.
Como se ha comentado, un exceso de haloperoxidasas puede inducir estrés halogenativo, de modo que estas enzimas han sido objeto de nuestro estudio. Primero se estudió la presencia en los pacientes de la TPO, peroxidasa que no solo es detectable en tiroides sino también en sangre y otros fluidos. Los resultados indican que el nivel medio de TPO está aumentado en el suero de los enfermos (1.736 ± 425 pg/ml en los pacientes versus 364 ± 212 pg/ml en controles; p < 0,05), lo que se debe a un aumento de la TPO sérica en el ∼35% de los pacientes estudiados (se considera un punto de corte o cut-off de 1.000 pg/ml, valor que no se alcanza en controles). Luego, se analizaron los niveles de LPO, haloperoxidasa que se detecta normalmente en sangre, leche y tejido cerebral. El LCR presentaba nivel elevado de LPO en el ∼43% de los pacientes estudiados (13,2 ± 1,4 ng/ml en los pacientes versus 8,5 ± 0,8 ng/ml en controles; p < 0,05; datos no publicados). Finalmente, se analizó el nivel de mieloperoxidasa en suero y LCR, pero no se detectaron diferencias respecto a controles. La tabla 1 muestra los niveles detectados de las haloperoxidasas mencionadas en suero y LCR.
Niveles de tiroperoxidasa, lactoperoxidasa y mieloperoxidasa, detectados en suero y LCR de enfermos con Parkinson, y sujetos control
| Parkinson | Controles | |
|---|---|---|
| TPO (pg/ml) | ||
| Suero | 1.736 ± 425* | 364±212 |
| LCR | Nd | Nd |
| LPO (ng/ml) | ||
| Suero | 765,8 ± 67 | 788,2 ± 65 |
| LCR | 13,2 ± 1,4* | 8,5 ± 0,8 |
| MPO (pg/ml) | ||
| Suero | 39.638 ± 6.950 | 29.202 ± 9.845 |
| LCR | 110 ± 28 | 91 ± 11 |
Media ± error estándar de la media.
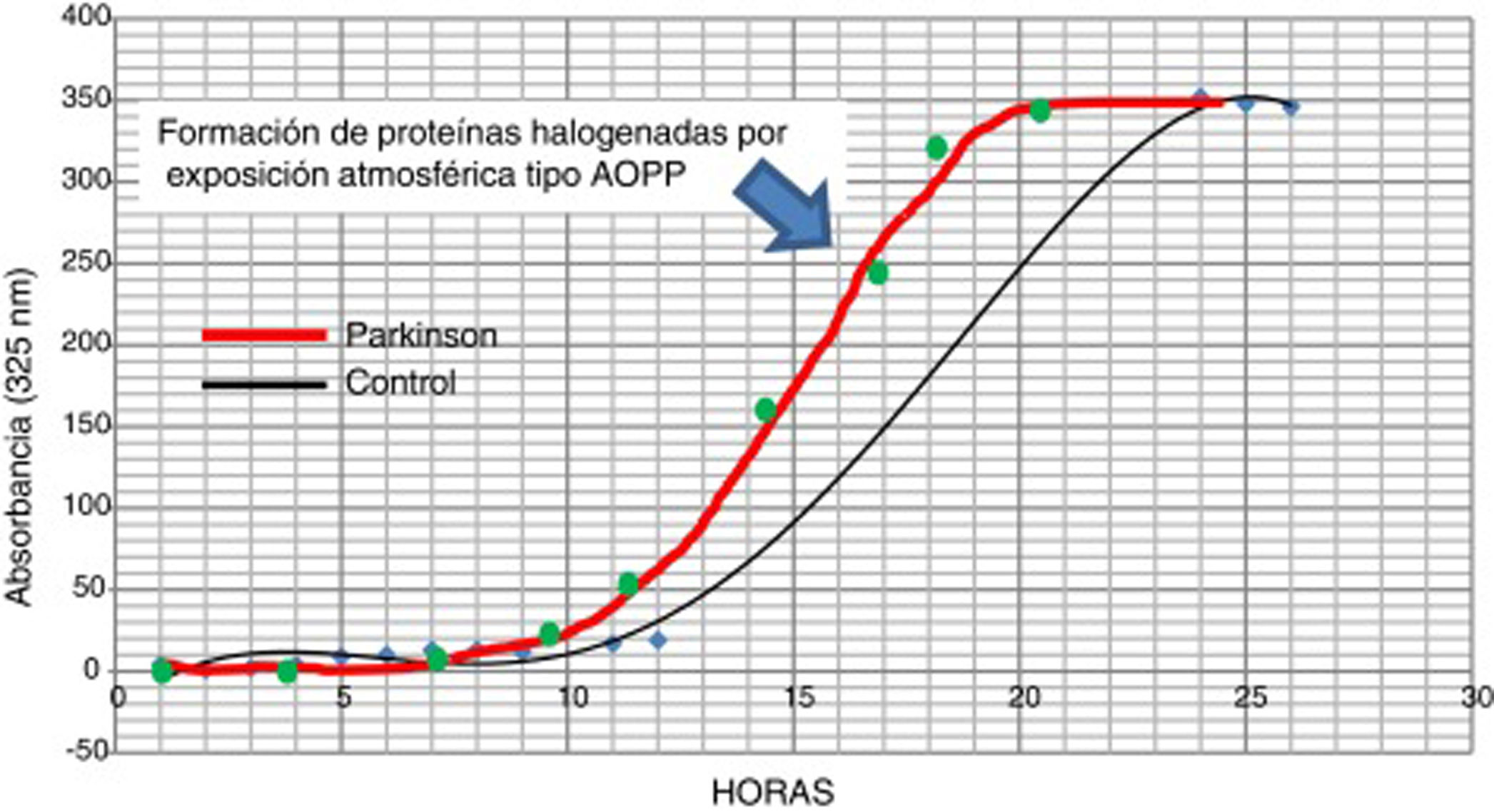
La posible presencia de estrés halogenativo en los enfermos se estudió en LCR por medio de curvas espectrofotométricas de halogenación espontánea atmosférica. Los fluidos se exponen a la atmósfera y se mide la presencia de derivados halogenados tipo AOPP a 325nm31,33. Como se observa en la figura 3, la halogenación del fluido sucede más rápidamente en el LCR de los enfermos que en los controles, pues la curva se desplaza a la izquierda. Esto indica que hay una «autohalogenación acelerada» en LCR de pacientes con EF26. Este hecho se observó en ∼50% de los enfermos analizados. No se ha detectado que el nivel de AOPP o de LPO se correlacione con «halogenación acelerada» del LCR en los enfermos.

Curvas de halogenación espontánea de líquido cefalorraquídeo por exposición atmosférica durante 27 horas, en 2 sujetos con niveles similares basales y de formación de AOPP. La curva gruesa es representativa de un paciente con enfermedad de Parkinson, y la fina de un sujeto control. Se observa el desplazamiento a la izquierda en el paciente, lo que indica una «autohalogenación acelerada» del LCR.
El hecho de que se haya detectado aumento en suero de TPO y proteínas halogenadas tipo AOPP, y exceso halogenativo e incremento de LPO y AOPP en LCR en un porcentaje de enfermos podría indicar que hay un incremento de productos halogenados amínicos en sangre y LCR, derivados de oxiácidos de halógenos. Como se ha comentado, entre los aminoácidos halogenados, aquellos que incluyen a la tirosina como clorotirosinas o yodotirosinas son de sumo interés para la EP, pues han demostrado toxicidad sobre neuronas de dopamina o son inhibidoras de la tirosina-hidroxilasa25,27,28. En nuestro laboratorio se ha estudiado en modelos celulares y animales la posible acción parkinsoniana» de la 3-yodo-L-tirosina. Pues bien, se ha detectado que dicha tirosina induce efectos parkinsonianos en modelos celulares y animales, con la aparición de inclusiones de αSYN y muerte de neuronas positivas a TH. El hecho de que nuestros resultados indiquen que la 3-yodo-L-tirosina tiene poder proagregante de αSYN realza su posible papel parkinsoniano, pues este hecho se considera un factor crucial en el desarrollo de la EP34,35. Estos resultados se han publicado recientemente29.
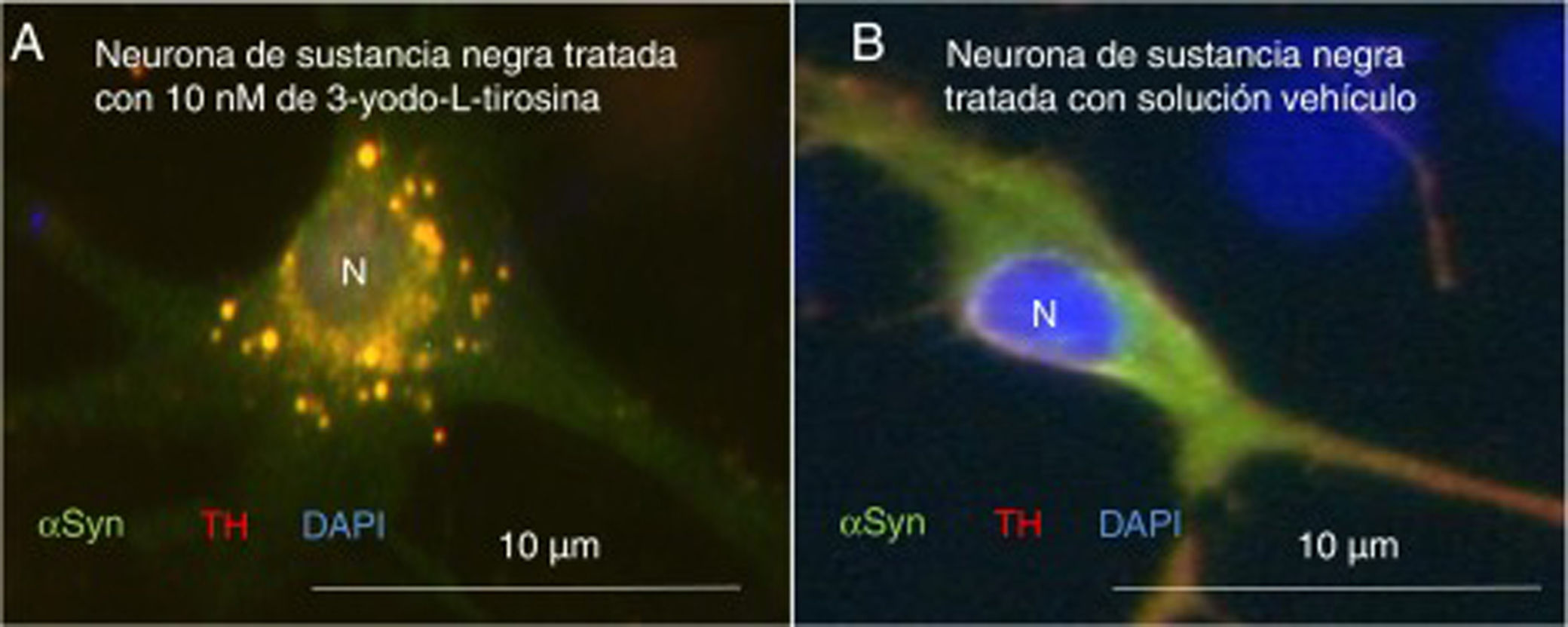
Como primer ejemplo de los resultados obtenidos, la exposición a 3-yodo-L-tirosina de neuronas de dopamina de la sustancia negra de ratón induce agregados intraneuronales que expresan αSYN y TH, como se observa en la figura 4.
 o solución vehículo (B), tras inmunocitoquímica para tirosina-hidroxilasa y α-sinucleína. A) Se observan numerosas inclusiones redondeadas que expresan tanto α-sinucleína (αSYN) como tirosina-hidroxilasa (TH), de ahí el color claro. Numerosos agregados se observan alrededor del núcleo. B) Se observa una neurona tratada con solución vehículo en la que hay señal clara difusa en el soma principalmente, y más oscura de TH en las neuritas. Es decir, la señal de αSYN se detecta difusa en el soma junto a TH, dando un color claro, y la señal de TH es más intensa en las neuritas. No se detectan inclusiones como en la figura A. El núcleo se tiñó con DAPI. N: núcleo. Fuente: Tomada de Frenández-Espejo26. Copyright © 2018, Fisiología, revista de la Sociedad Española de Ciencias Fisiológicas.")
Neuronas dopaminérgicas de sustancia negra en cultivo tratadas con 10μM de 3-yodo-L-tirosina (A) o solución vehículo (B), tras inmunocitoquímica para tirosina-hidroxilasa y α-sinucleína. A) Se observan numerosas inclusiones redondeadas que expresan tanto α-sinucleína (αSYN) como tirosina-hidroxilasa (TH), de ahí el color claro. Numerosos agregados se observan alrededor del núcleo. B) Se observa una neurona tratada con solución vehículo en la que hay señal clara difusa en el soma principalmente, y más oscura de TH en las neuritas. Es decir, la señal de αSYN se detecta difusa en el soma junto a TH, dando un color claro, y la señal de TH es más intensa en las neuritas. No se detectan inclusiones como en la figura A. El núcleo se tiñó con DAPI.
N: núcleo.
Fuente: Tomada de Frenández-Espejo26. Copyright © 2018, Fisiología, revista de la Sociedad Española de Ciencias Fisiológicas.
También en un modelo de inyección intracerebral unilateral en ratones, la inyección intraestriatal de 3-yodo-L-tirosina induce daño del circuito nigroestriado. La densidad TH del estriado se reduce un ∼30% tras las inyecciones y el número de neuronas TH+de la sustancia negra cae ∼35%. Esto se acompaña de alteraciones comportamentales «parkinsonianas» en ratones, como giro inducido o acinesia/bradicinesia.
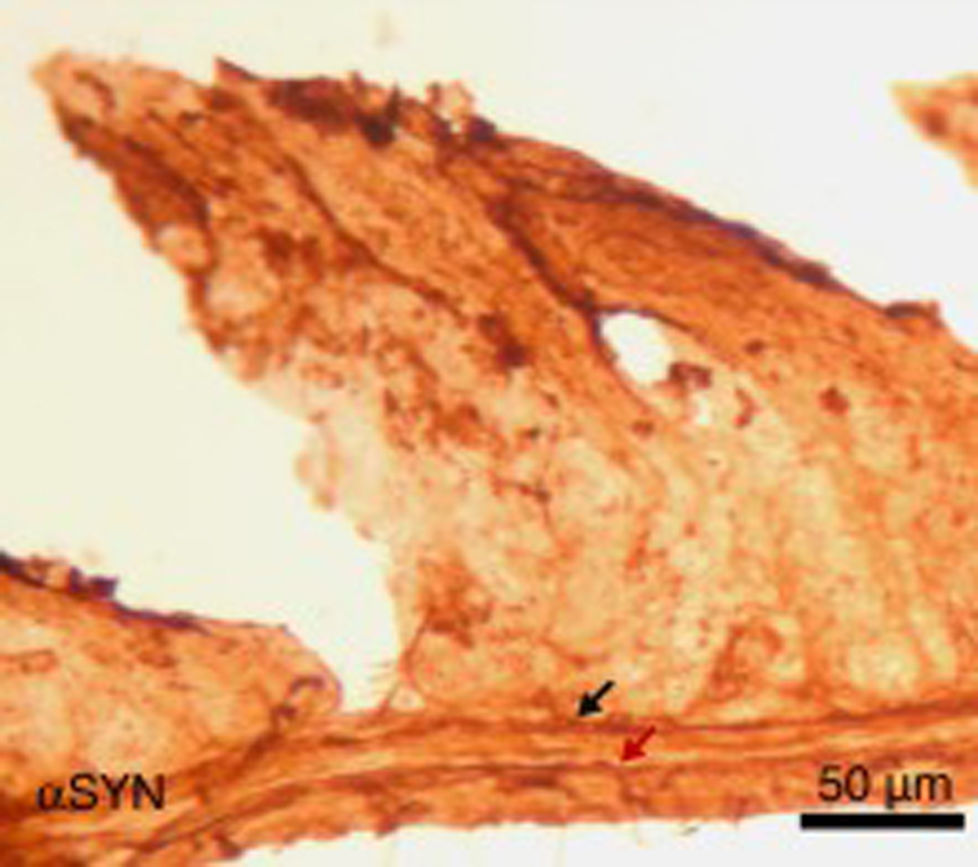
Finalmente, se ha estudiado, en ratones, el efecto de la inyección intraperitoneal repetida de 3-yodo-L-tirosina sobre la pared yeyunal. Hay que decir que los agregados de αSYN y la degeneración de neuronas positivas a TH se detectan en humanos en otras localizaciones periféricas al sistema nervioso central: destaca el sistema nervioso entérico del tracto digestivo, desde el esófago hasta el recto34,36,37. Como se observa en la figura 5, la inyección repetida de 10μM de 3-yodo-L-tirosina induce la presencia de αSYN agregada en los plexos de Auerbach y Meissner, con engrosamiento de las fibras nerviosas, lo que no se observa en sujetos control tratados con vehículo. También hay degeneración de neuronas TH+y sus fibras. Estos resultados encajan también con estudios con modelos experimentales de EP como el de MPTP. Así, se sabe que el MPTP induce un decremento intestinal de 40-80% de neuronas TH+en ratones, con la presencia de agregados de αSYN en los plexos intramurales del sistema nervioso entérico de ratones38-40.
, en ratones tratados intraperitonealmente con 4 inyecciones semanales de 10μM de 3-yodo-L-tirosina. Se observan los plexos de Meissner y Auerbach (flechas) positivos a αSYN, engrosados y con aglomerados de la proteína. Este hecho no se observa en ratones control, en los que los plexos son finos y expresan poco αSYN.")
Imagen de la pared yeyunal tras inmunohistoquímica de α-sinucleína (αSYN), en ratones tratados intraperitonealmente con 4 inyecciones semanales de 10μM de 3-yodo-L-tirosina. Se observan los plexos de Meissner y Auerbach (flechas) positivos a αSYN, engrosados y con aglomerados de la proteína. Este hecho no se observa en ratones control, en los que los plexos son finos y expresan poco αSYN.
Los estudios en el laboratorio han permitido identificar cambios del metabolismo de halógenos en suero y LCR de pacientes con EF que indican la presencia de «autohalogenación acelerada» del LCR de los pacientes o aumento de haloperoxidasas, enzimas de síntesis de oxiácidos de halógenos, en concreto, TPO sérica o LPO del LCR. Además, se ha detectado un exceso en suero de algunas moléculas derivadas de exceso de halogenación, como proteínas con halogenación avanzada tipo AOPP. Estos hechos, autohalogenación acelerada e incremento de haloperoxidasas y proteínas AOPP, indican la presencia de estrés halogenativo en la EP. Además, la 3-yodo-L-tirosina, un derivado halogenado, muestra toxicidad parkinsoniana en modelos experimentales, pues se ha observado que induce agregados de α-SYN y daño de las neuronas de dopamina en cerebro e intestino en ratones.
La hipótesis que se maneja es que en la EP existe un exceso halogenativo, relacionado con una alteración enzimática de síntesis o degradación de oxiácidos de halógenos y sus derivados halogenados. Es decir, como proceso patológico en la enfermedad, tendría lugar una haloenzimopatía, que originaría estrés halogenativo que podría relacionarse con el daño del sistema nervioso.
FinanciaciónEl trabajo de investigación ha sido subvencionado por la Consejería de Economía, Conocimiento, Empresas y Universidad, Junta de Andalucía (ref. BIO127) y la Sociedad Andaluza de Neurología (ref. SUBAIA2015/006).
Conflicto de interesesEl autor declara no tener ningún conflicto de intereses.
El autor agradece ante todo la colaboración de los enfermos y controles que han participado en los estudios. Todos los participantes han dado su consentimiento informado y los estudios han sido aprobados por los comités éticos correspondientes. El autor agradece la ayuda y el apoyo de Cristian Bis-Humbert y Silvia Castellano (Universidad de Sevilla), José Manuel García Moreno (Hospital Macarena de Sevilla), ÿngel Martín de Pablos (Hospital Macarena de Sevilla y Universidad de Sevilla), José Chacón Peña (Hospital Quirón Infanta Luisa de Sevilla) y Fátima Damas Hermoso (Hospital de Valme de Sevilla), Isaac Túnez Fiñana (Universidad de Córdoba) y Adrián Fernández (Hospital Santa Isabel de Sevilla).

