




Laboratory studies identified changes in the metabolism of halogens in the serum and cerebrospinal fluid (CSF) of patients with Parkinson’s disease, which indicates the presence of “accelerated self-halogenation” of CSF and/or an increase in haloperoxidases, specifically serum thyroperoxidase and CSF lactoperoxidase. Furthermore, an excess of some halogenated derivatives, such as advanced oxygenation protein products (AOPP), has been detected in the CSF and serum. “Accelerated self-halogenation” and increased levels of haloperoxidases and AOPP proteins indicate that halogenative stress is present in Parkinson’s disease. In addition, 3-iodo-L-tyrosine, a halogenated derivative, shows “parkinsonian” toxicity in experimental models, since it has been observed to induce α-synuclein aggregation and damage to dopaminergic neurons in the mouse brain and intestine. The hypothesis is that patients with Parkinson’s disease display halogenative stress related to a haloenzymatic alteration of the synthesis or degradation of oxyacid of halogens and their halogenated derivatives. This halogenative stress would be related to nervous system damage.
Los estudios en el laboratorio han permitido identificar cambios del metabolismo de halógenos en suero y líquido cefalorraquídeo (LCR) de pacientes con enfermedad de Parkinson, que indican la presencia de «autohalogenación acelerada» del LCR de los pacientes o aumento de haloperoxidasas, en concreto, tiroperoxidasa en sangre y lactoperoxidasa en LCR. Además, se ha detectado un exceso en suero y LCR de algunos derivados halogenados, como proteínas con halogenación avanzada tipo advanced oxidation protein products (AOPP). Estos hechos, «autohalogenación acelerada» e incremento de haloperoxidasas y proteínas AOPP, indican la presencia de estrés halogenativo en la enfermedad de Parkinson. Además, un derivado halogenado, la 3-yodo-L-tirosina, muestra toxicidad parkinsoniana en modelos experimentales, pues se ha observado que induce agregados de α-sinucleína y daño de las neuronas de dopamina en cerebro e intestino en ratones. La hipótesis que se maneja es que en la enfermedad de Parkinson existe un exceso halogenativo, relacionado con una alteración haloenzimática de síntesis o degradación de oxiácidos de halógenos y sus derivados halogenados. Este estrés halogenativo se relacionaría con el daño del sistema nervioso.
Oxidative stress is defined as an imbalance between the production of reactive oxygen species and the antioxidant system, and is currently thought to be an important pathogenic mechanism in Parkinson’s disease (PD).1,2 The different types of oxidative stress3 include peroxidative stress, oxidative stress due to reactive nitrogen species (e.g., nitric oxide [•NO]), and stress due to halogen species (e.g., hypochlorous acid [HOCl]).
Peroxidative stress is caused by excessive superoxide anion (•O2−) or hydrogen peroxide (H2O2), and can be detected in the brain tissue, blood, and cerebrospinal fluid (CSF) of patients with PD. The substantia nigra is known to undergo intense peroxidative stress, showing a considerable increase in levels of such oxidative markers as peroxidised lipids,4 8-hydroxyguanosine (a marker of oxidative stress to DNA),5 carbonylated proteins,6 and advanced lipoxidation end products.7 CSF studies show significantly reduced activity of numerous antioxidant enzymes related to peroxidation in patients with PD.8
Excessive •NO activity induces 2 types of stress: nitrative stress and S-nitrosylation. In nitration, protein oxidation occurs in tyrosine residues, whereas S-nitrosylation involves cysteine residues. In patients with PD, nitrative stress affects proteins that are highly relevant in the disease, such as manganese superoxide dismutase, tyrosine hydroxylase (TH), and α-synuclein (αSYN). Nitrated manganese superoxide dismutase is detected in Lewy bodies and in the CSF of these patients.9 Nitrated superoxide dismutase presents loss of function and causes mitochondrial vacuolation and lipid peroxidation in mice; these phenomena are observed in PD.10 TH is selectively nitrated in PD, inhibiting its enzymatic function; this may play a pathogenic role.11,12 The protein 3-nitrotyrosine αSYN (3NT-SYN) is a component of Lewy bodies, and represents an anomalous form of αSYN that is also involved in PD pathogenesis.13,14 Recent studies analysing the serum of patients with PD detected nitrative stress, indicated by significant increases in 3-nitrotyrosine and 3NT-αSYN proteins.15 Finally, in S-nitrosylation, nitric oxide binds to cysteine thiols, giving rise to nitrosothiol derivatives. This modifies the functionality of numerous proteins involved in PD, such as protein disulfide-isomerase and parkin.16,17 When they are nitrosylated, both proteins lose their antioxidant and ubiquitination effects, facilitating protein aggregation and deposition.18,19
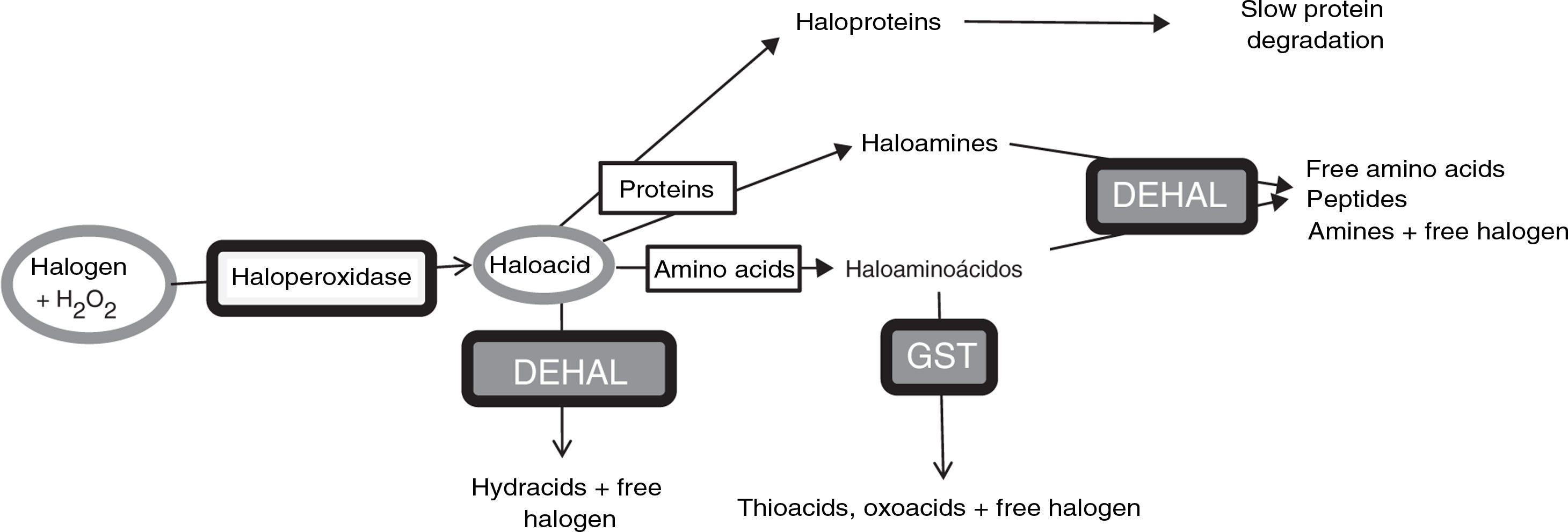
Halogenative stress and Parkinson’s diseaseHalogenative stress is caused by an excess of reactive halogen species, mainly such halogen oxoacids as hypochlorous acid and hypoiodous acid. Haloperoxidases are enzymes that catalyse the conversion of H2O2 into oxoacids through the incorporation of halogens (Fig. 1).20–23 Halogen species are increasingly important in understanding the aetiology of PD and other neurodegenerative diseases.1,3,24–26 Important examples of haloperoxidases are those expressed in white blood cells and microglia, such as myeloperoxidase and eosinophil peroxidase, and in cells presenting iodine/bromine uptake (e.g., cells from the thyroid, salivary glands, or breast), which include thyroid peroxidase (TPO), salivary peroxidase, and lactoperoxidase (LPO). Oxoacids, in turn, halogenate proteins and amino acids, increasing the levels of these derivatives when there is an excess of oxoacids or haloperoxidase activity.

Metabolic pathways associated with halogenative stress and the proteins and amino acids involved. Haloperoxidases produce oxoacids from halogens and hydrogen peroxide. Oxoacids are degraded to hydracids by the action of dehalogenases, or may halogenate proteins and amino acids. Proteins are converted into haloproteins or haloamines, and amino acids are converted into haloamino acids. Haloproteins degrade slowly. Haloamines and haloamino acids are degraded by dehalogenases, generating amino acids, peptides, and amines; and by glutathione-S-transferase, generating thioacids and oxoacids. Therefore, excessive haloperoxidase activity or a defect of enzymatic degradation may lead to halogenative stress, with elevated levels of oxoacids and halogenated derivatives. DEHAL: dehalogenases; GST: glutathione-S-transferase; H2O2: hydrogen peroxide.
Halogenated amino acids derived from tyrosine, such as chlorotyrosines and iodotyrosines, are of great interest in the study of PD, as they present dopaminergic neurotoxicity and inhibit tyrosine hydroxylase.1,25,27–29 Other chloroamino acids, such as chlorocysteine and chlorolysine, also damage dopaminergic neurons by altering membrane proteins.30 Key members of the family of halogenated proteins are hypochlorite-modified proteins, haloamines, and advanced oxidation protein products (AOPP).31 Hypochlorite-modified proteins present intense chlorination in cysteine residues. Haloamines are halogenated oligopeptides. AOPPs are anomalous proteins presenting strong halogenation, and particularly chlorination, of dityrosine or lysine residues.
A study conducted at our laboratory detected increased serum TPO concentration in approximately 35% of patients with PD15 and increased CSF LPO concentration in approximately 43% (unpublished data). These patients often present elevated AOPP levels in serum and CSF.15 All these findings indicate that patients with PD present halogenative stress. Degradation of halogen oxoacids and their amine derivatives (haloamino acids and haloamines) is mainly catalysed by dehalogenases (DEHAL), although glutathione-S-transferase is also involved. Iodotyrosine dehalogenase 1 (DEHAL 1) is an important member of this group of enzymes. This protein presents 2 main isoforms (DEHAL 1 and DEHAL 1B); these oxidoreductase enzymes are encoded by the IYD gene in humans.32 Halogen protein derivatives, which usually appear in the context of prolonged situations of halogenative stress (e.g., chronic inflammation or metabolic disorders), degrade slowly and remain present in bodily fluids.31Fig. 1 shows the metabolic and halogenative stress pathways described above.
Anomalous halogenative activity in Parkinson’s diseaseLaboratory studies have identified halogenation changes in the serum and CSF of patients with PD. First, elevated AOPP levels were detected in the serum. Serum AOPP levels also correlate with the length of time that the disease remains at Hoehn and Yahr stage 2 without passing to more advanced stages (Fig. 2). Patients at stage 2 with over 13 years’ disease progression (i.e., those with relatively good motor status) present significantly lower serum AOPP levels. These patients may have more effective antihalogenative mechanisms or less severe halogenative stress. No relevant changes were detected in serum or CSF levels of myeloperoxidase, the haloperoxidase with greatest involvement in AOPP production.25
![Relationship between serum levels of advanced oxidation protein products and duration of Parkinson’s disease. All patients (n=34) were classed as stage 1 or 2 on the Hoehn and Yahr scale. Patients with disease duration longer than 13 years present low serum levels of advanced oxidation protein products (AOPP) (rhombi: duration < 13 years; mean [standard error of the mean] AOPP, 487 [60] μM; squares: duration > 13 years; mean AOPP, 87 [9] μM; P<.01 [t test]). Continuous lines show the mean value for each patient group. The Hoehn and Yahr scale is commonly used to characterise symptom progression in Parkinon’s disease, with scores ranging from 1 (unilateral involvement only) to 5 (confinement to bed or wheelchair unless assisted). Source: modified from García-Moreno et al.25](https://static.elsevier.es/multimedia/21735808/0000003700000008/v2_202210110613/S2173580821000195/v2_202210110613/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9idGjqO6E2fFf084sfhX5KtshEksBDze+4mYfOh7kmT462Z1K8JmJ0fq6XWL+AyZL7znrYlt1ogIS/fqJNdEVPFh92Z7zuHKU7vG27+fQmpFi6/zwSa5ZDfhxNMaxJjOF+GvKKO9XF8fiH+eN2p2+nc1BjWXooY/IYxRgbf3VfF0SM/I0NOOwmbVigFVjQ4xk+Ocmf5hdank6zkwmuNs6EsDLtxZuoRpgaVhXu/C16fVI= "Relationship between serum levels of advanced oxidation protein products and duration of Parkinson’s disease. All patients (n=34) were classed as stage 1 or 2 on the Hoehn and Yahr scale. Patients with disease duration longer than 13 years present low serum levels of advanced oxidation protein products (AOPP) (rhombi: duration < 13 years; mean [standard error of the mean] AOPP, 487 [60] μM; squares: duration > 13 years; mean AOPP, 87 [9] μM; P<.01 [t test]). Continuous lines show the mean value for each patient group. The Hoehn and Yahr scale is commonly used to characterise symptom progression in Parkinon’s disease, with scores ranging from 1 (unilateral involvement only) to 5 (confinement to bed or wheelchair unless assisted). Source: modified from García-Moreno et al.25")
Relationship between serum levels of advanced oxidation protein products and duration of Parkinson’s disease. All patients (n=34) were classed as stage 1 or 2 on the Hoehn and Yahr scale. Patients with disease duration longer than 13 years present low serum levels of advanced oxidation protein products (AOPP) (rhombi: duration < 13 years; mean [standard error of the mean] AOPP, 487 [60] μM; squares: duration > 13 years; mean AOPP, 87 [9] μM; P<.01 [t test]). Continuous lines show the mean value for each patient group. The Hoehn and Yahr scale is commonly used to characterise symptom progression in Parkinon’s disease, with scores ranging from 1 (unilateral involvement only) to 5 (confinement to bed or wheelchair unless assisted). Source: modified from García-Moreno et al.25
Furthermore, AOPPs were observed in the CSF of patients with PD, whereas they were undetectable in controls. AOPPs were detected in the CSF of approximately 53% of patients, with a mean (standard error of the mean [SEM]) AOPP concentration of 11.4 (2) μM. Hoehn and Yahr stage and progression time were not correlated with this marker; however, CSF positivity for AOPPs clearly indicates halogenative stress in the central nervous system.
As discussed above, excess haloperoxidase levels may induce halogenative stress, hence the selection of these enzymes as the subject of analysis. We initially studied levels of TPO, which is usually detectable in the blood and other fluids, as well as in the thyroid gland. Our findings indicate that the mean (SEM) serum TPO concentration is higher in patients with PD than in controls (1736 [425] pg/mL vs 364 [212] pg/mL; P<.05), which was explained by elevated serum TPO levels in approximately 35% of the patients studied (the cut-off point established was 1000pg/mL, which was not reached in controls). We subsequently analysed levels of LPO, a haloperoxidase that is usually detected in blood, breast milk, and brain tissue. CSF LPO concentration was elevated in approximately 43% of patients studied (13.2 [1.4] ng/mL in patients and 8.5 [0.8] ng/mL in controls; P<.05; unpublished data). Finally, we analysed serum and CSF levels of myeloperoxidase; no significant differences were observed. Table 1 shows levels of these haloperoxidases in the serum and CSF.
Levels of thyroperoxidase, lactoperoxidase, and myeloperoxidase in the serum and CSF of patients with Parkinon’s disease and healthy controls.
| Patients | Controls | |
|---|---|---|
| TPO, pg/mL | ||
| Serum | 1736 (425)* | 364 (212) |
| CSF | Ud | Ud |
| LPO, ng/mL | ||
| Serum | 765.8 (67) | 788.2 (65) |
| CSF | 13.2 (1.4)* | 8.5 (0.8) |
| MPO, pg/mL | ||
| Serum | 39638 (6950) | 29202 (9845) |
| CSF | 110 (28) | 91 (11) |
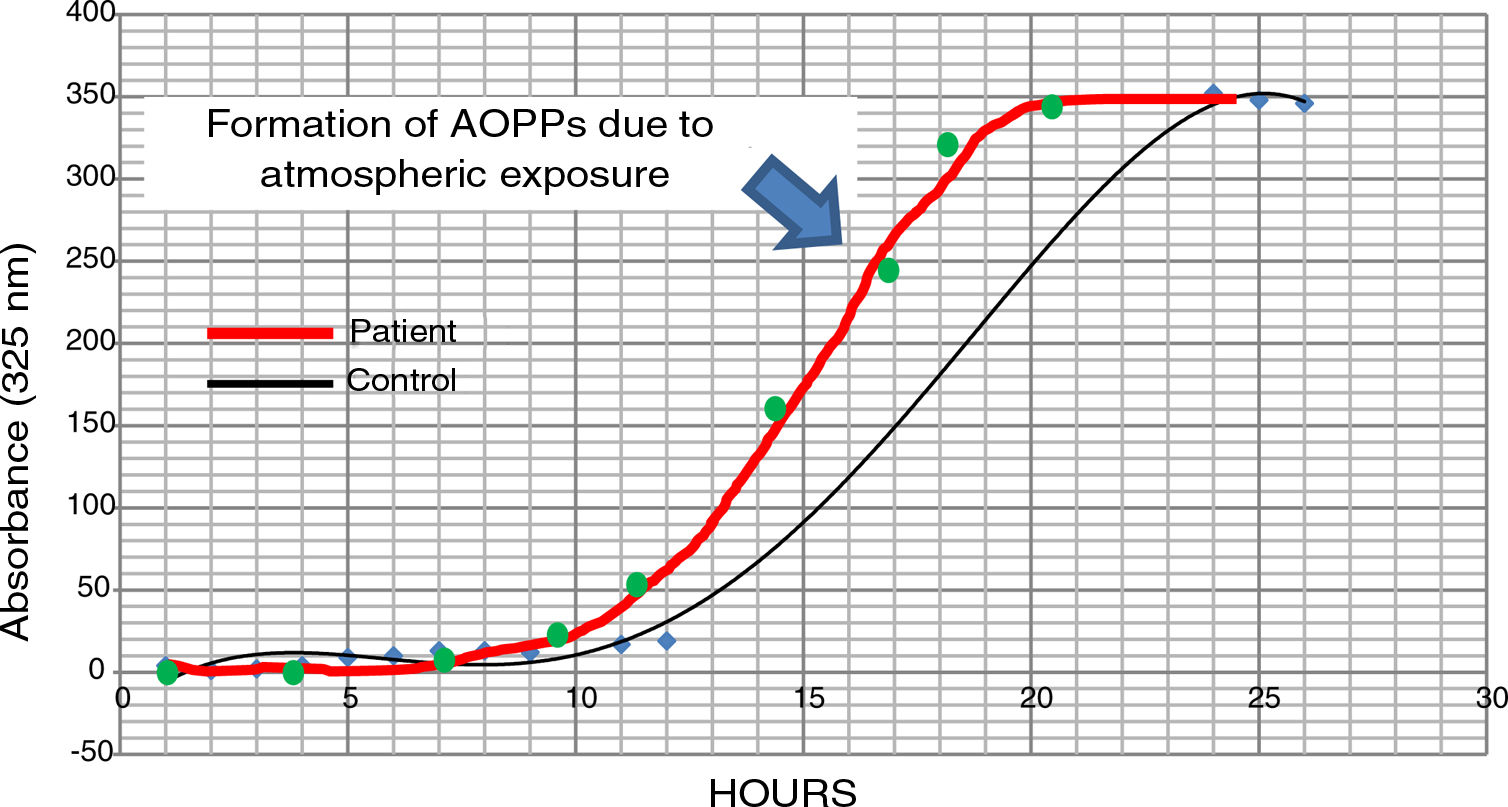
The possible presence of halogenative stress in the CSF of patients with PD was studied using spectrophotometry curves for spontaneous halogenation due to atmospheric exposure. Samples were exposed to the atmosphere and the presence of AOPPs was measured at 325nm.31,33 As shown in Fig. 3, halogenation of CSF is more rapid in samples from patients than in samples from controls, with the curve displaced to the left. This indicates an “accelerated self-halogenation” effect in the CSF of patients with PD.26 These findings were observed in approximately 50% of the patients analysed. Levels of AOPPs and LPO were not correlated with accelerated halogenation in the CSF of patients with PD.

Spectrophotometry curves for spontaneous halogenation of cerebrospinal fluid due to atmospheric exposure for 27hours; results from 2 individuals with similar baseline levels and formation of advanced oxidation protein products. The bold line represents data from a patient with Parkinson’s disease, and the fine line shows data from a healthy control. Data from the patient is displaced to the left, indicating accelerated self-halogenation.
The fact that elevated serum TPO and AOPP levels, excessive halogenation, and elevated CSF LPO and AOPP levels have been detected in some patients with PD may suggest increased presence in the blood and CSF of halogenated amine products derived from halogen oxoacids. As previously discussed, halogenated amino acids derived from tyrosine, such as chlorotyrosines and iodotyrosines, are of great interest in the study of PD, as they present toxicity to dopaminergic neurons and inhibit TH.25,27,28 At our laboratory, we have studied the possible parkinsonian action of 3-iodo-L-tyrosine in cellular and animal models. Our findings suggest that this molecule induces parkinsonian effects in cellular and animal models, with the appearance of αSYN inclusions and the death of TH-positive neurons. The fact that 3-iodo-L-tyrosine seems to promote αSYN aggregation highlights its potential parkinsonian effects, as this mechanism is considered a crucial factor in PD pathogenesis.34,35 These results were published in a recent study.29
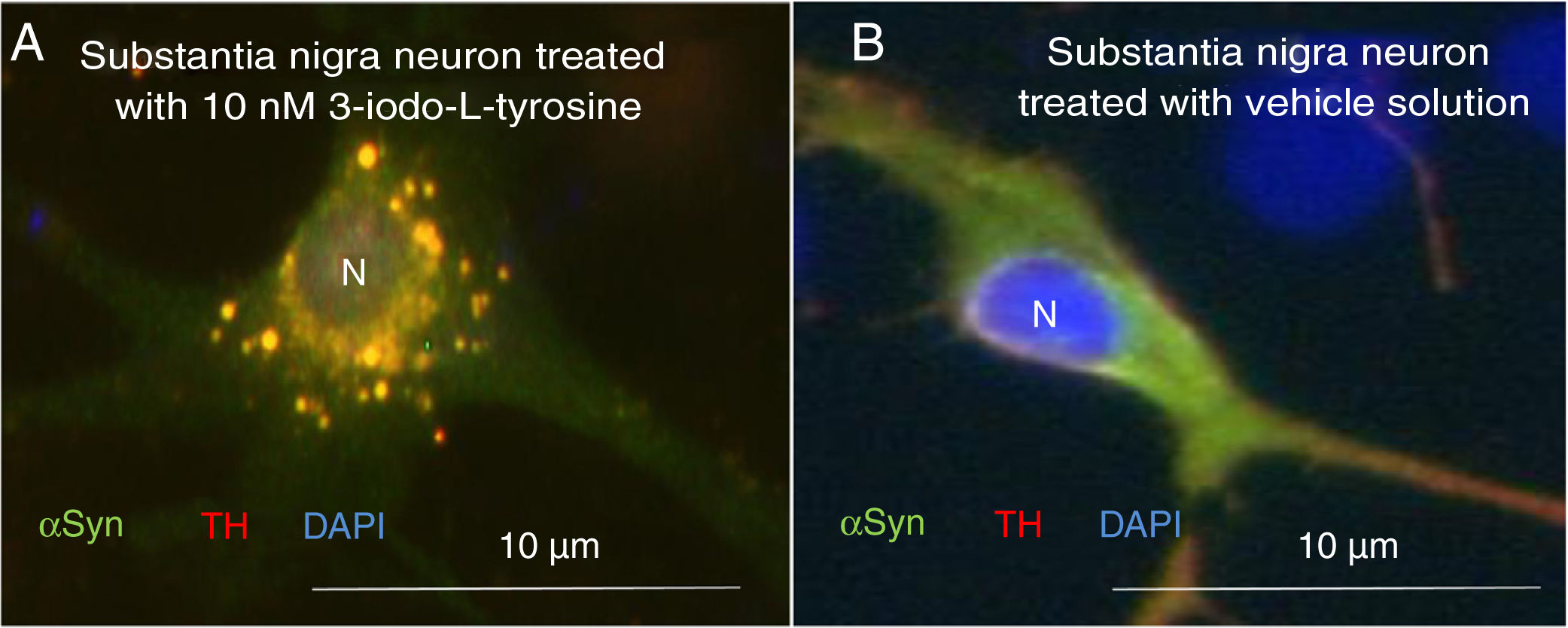
Exposure to 3-iodo-L-tyrosine induced the formation of intraneuronal aggregates expressing αSYN and TH in dopaminergic neurons from the mouse substantia nigra (Fig. 4).
 and α-synuclein (αSYN) in a culture of dopaminergic neurons from the substantia nigra, treated with 10μM of 3-iodo-L-tyrosine (A) or vehicle solution (B). (A) The cells present numerous round inclusions expressing both αSYN and TH, hence the light colouration. Numerous aggregates are observed surrounding the nucleus. (B) A neuron treated with vehicle solution, displaying diffuse light signal mainly in the soma, with darker signal for TH in the neurites. In other words, the expression of both proteins is diffuse in the soma, with TH expression being more intense in the neurites. The sample does not present inclusions like those shown in panel A. The nucleus was stained with DAPI. N: nucleus. Source: taken from Fernández-Espejo.26 Copyright © 2018, Fisiología, journal of the Spanish Society of Physiological Sciences.")
Immunocytochemistry images showing the distribution of tyrosine hydroxylase (TH) and α-synuclein (αSYN) in a culture of dopaminergic neurons from the substantia nigra, treated with 10μM of 3-iodo-L-tyrosine (A) or vehicle solution (B). (A) The cells present numerous round inclusions expressing both αSYN and TH, hence the light colouration. Numerous aggregates are observed surrounding the nucleus. (B) A neuron treated with vehicle solution, displaying diffuse light signal mainly in the soma, with darker signal for TH in the neurites. In other words, the expression of both proteins is diffuse in the soma, with TH expression being more intense in the neurites. The sample does not present inclusions like those shown in panel A. The nucleus was stained with DAPI. N: nucleus. Source: taken from Fernández-Espejo.26 Copyright © 2018, Fisiología, journal of the Spanish Society of Physiological Sciences.
Unilateral injection of 3-iodo-L-tyrosine into the striatum in mice induced damage to the nigro-striatal pathway. The density of TH in the striatum decreased by approximately 30% after the injections, and the number of TH-positive neurons decreased by approximately 35%. The animals also displayed “parkinsonian” behavioural alterations, such as induced spinning behaviour and akinesia/bradykinesia.
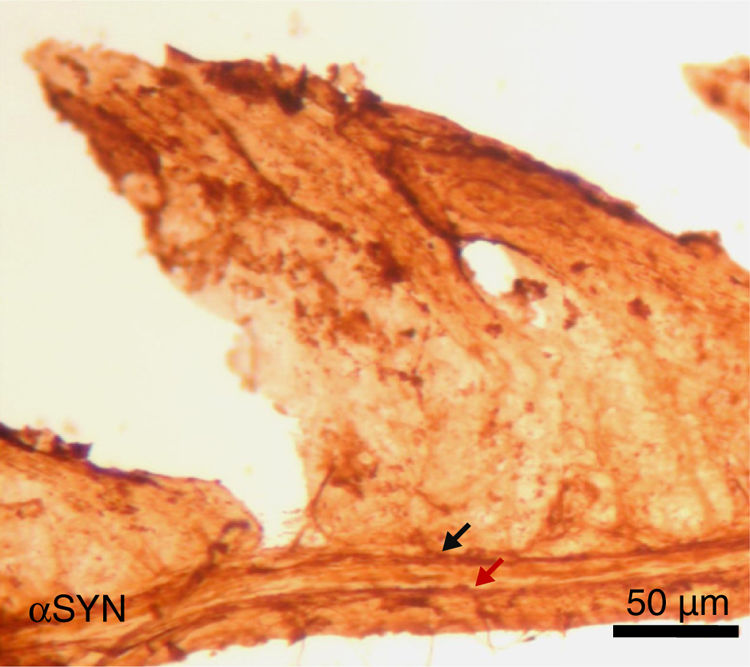
Finally, the effect of repeated intraperitoneal injection of 3-iodo-L-tyrosine on the jejunal wall was studied in mice. In humans, αSYN aggregates and degradation of TH-positive neurons are also detected in other peripheral locations of the nervous system, such as in the enteric nervous system, from the oesophagus to the rectum.34,36,37 As is shown in Fig. 5, repeated injection of 10μM of 3-iodo-L-tyrosine induces αSYN aggregation in the Auerbach plexus and the Meissner plexus, with thickening of nerve fibres; this change was not observed in control subjects injected with vehicle solution. Degeneration of TH-positive neurons and their fibres is also observed. These results are consistent with those reported for experimental models of PD, such as MPTP. MPTP is known to cause a loss of 40%–80% of TH-positive neurons in the intestines of mice, as well as the appearance of αSYN aggregates in intramural plexi in the enteric nervous system.38–40
; sample taken from a mouse treated with 4 weekly intraperitoneal injections of 10μM 3-iodo-L-tyrosine. The Meissner plexus and Auerbach plexus (arrows) are positive for αSYN, and present thickening, with aggregates of the protein. These findings were not observed in control mice, in which these plexi were thin and expressed little αSYN.")
Image of the jejunal wall after immunohistochemistry staining for α-synuclein (αSYN); sample taken from a mouse treated with 4 weekly intraperitoneal injections of 10μM 3-iodo-L-tyrosine. The Meissner plexus and Auerbach plexus (arrows) are positive for αSYN, and present thickening, with aggregates of the protein. These findings were not observed in control mice, in which these plexi were thin and expressed little αSYN.
Laboratory studies have identified changes in the metabolism of halogens in the serum and CSF of patients with PD, suggesting a process of “accelerated self-halogenation” in the CSF and increased levels of haloperoxidase enzymes, which synthesise halogen oxoacids, and specifically TPO in the serum and LPO in the CSF. Serum analysis has also shown elevated levels of some molecular derivatives of excessive halogenation, including AOPPs. The accelerated self-halogenation process observed and the increased levels of haloperoxidases and AOPPs indicate that halogenative stress is present in PD. Furthermore, 3-iodo-L-tyrosine, a halogen derivative, has shown parkinsonian toxicity in experimental models, inducing αSYN aggregation and damage to dopaminergic neurons in the mouse brain and intestine.
Based on this evidence, we believe that excess halogenation occurs in PD due to alterations to enzymes responsible for synthesising or degrading halogen oxoacids and their halogenated derivatives. In other words, one of the processes involved in PD pathogenesis may be a halo-enzymopathy that would result in halogenative stress-induced damage to the nervous system.
FundingThis study received funding from the Regional Ministry of Economy, Knowledge, Enterprise, and Universities of the regional government of Andalusia (ref. BIO127) and the Andalusian Society of Neurology (ref. SUBAIA2015-006).
Conflicts of interestThe author has no conflicts of interest to declare.
The author would like to thank all the patients and controls who participated in the study. All participants gave informed consent and the studies were approved by the relevant ethics committees. The author is also grateful for the support of Cristian Bis-Humbert and Silvia Castellano (Universidad de Sevilla), José Manuel García Moreno (Hospital Virgen Macarena, Seville), Ángel Martín de Pablos (Hospital Virgen Macarena, Seville, and Universidad de Sevilla), José Chacón Peña (Hospital Quirón Infanta Luisa, Seville), Fátima Damas Hermoso (Hospital Virgen de Valme, Seville), Isaac Túnez Fiñana (Universidad de Córdoba), and Adrián Fernández (Clínica Santa Isabel, Seville).
Please cite this article as: Fernández-Espejo E. ¿Presenta la enfermedad de Parkinson una haloenzimopatía? Neurología. 2022;37:661–667.
