




La secuenciación exómica elucida síndromes genéticos hasta en un 25% de los individuos con condiciones genéticas sin diagnóstico posterior a múltiples estudios genéticos1,2. Recientemente (2013), Bainbridge y Ropers reportaron variantes de novo por mutaciones truncas en el gen Additionale sex combs-like 3 (ASXL3, locus 18q12.2, 12 exones) en 4 pacientes con retraso psicomotor grave, dificultad en el aprendizaje y para la alimentación3.
Se evaluó a un masculino de 3 años de edad, cuarto hijo de padres sanos, no consanguíneos y mexicanos con 3 hermanos sanos. Posterior a un embarazo normal, nació vía vaginal a término (longitud: p>97; peso: p>85; OFC: p97; Apgar: 7,9). Alcanzó sostén cefálico a los 6 meses y la sedestación al año. A los 36 meses se observa retraso del desarrollo neurológico grave, sin lenguaje y marcha. Cursa con microcefalia (−4,35 DE), talla baja (−3,5 DE) y pobre ganancia de peso (−4,6 DE). El fenotipo facial incluye frente prominente, hipertelorismo, implantación anterior del cabello alta, cejas delgadas, ligera sinofridia, narinas antevertidas con hipoplasia de alas nasales, paladar arqueado y orejas prominentes de implantación baja. También se observan almohadillas dactilares, primer dedo ancho en manos y en pies y criptorquidia (fig. 1). LA IRM cerebral demostró atrofia cortical. El cariotipo (46,XY) y la hibridación genómica comparativa (400k) fueron normales. En Baylor Miraca Genetics Laboratories se realizó secuenciación exómica completa a partir del DNA genómico purificado de sangre periférica del paciente. Se realizó fragmentación por sonicación y ligamiento con adaptadores de secuencia multiplex de la plataforma Illumina4. El proceso de mejora/captura se obtuvo hibridando el microarreglo personalizado por NimbleGen (VCRome 2.1)5, así como sondas adicionales para 3,650 genes mendelianos y de genoma mitocondrial6. El análisis se realizó con el sistema Illumina HiSeq 2000 (cobertura media exómica del 95% y nucleotídica >100X). El llamado de variantes y las anotaciones se realizaron utilizando Atlas-SNP/SNP-anno y Atlas-indel/HGSC-anno desarrolladas internamente por Baylor College of Medicine. Las variantes de novo y las predicciones in silico para cambios sin sentido se reportaron de acuerdo al American College of Medical Genetics7. Se detectó una deleción de 4pb en el cromosoma 18:31320359, en forma heterocigota, NM_030632.2 (ASXL3): c.2992-2995del, p.(E998fs) en el exón 11, el cual causa un cambio en el marco de lectura que produce una proteína aberrante que se predice deletérea. La secuenciación Sanger del gen ASXL3 en los padres fue normal por lo que se infiere que la variante fue de novo en el paciente.
 Frente amplia, cresta metópica prominente, boca permanentemente abierta y orejas de implantación baja con rotación posterior. C) Almohadillas dactilares. D y E) Dedos anchos, especialmente el primer dedo y ortejo.")
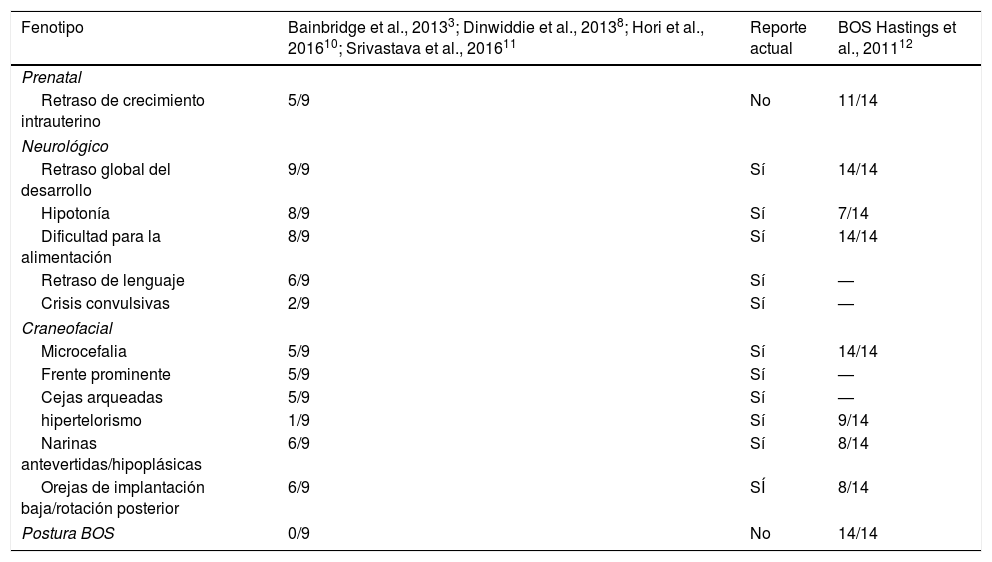
Hasta la fecha se describen pocos pacientes con el BRS (MIM: 615485). El fenotipo sutil y variable deriva en que el diagnóstico se establece con la secuenciación completa del genoma o del exoma3,8–11. Las características clínicas comunes, retraso psicomotor grave, dismorfias faciales, falla del crecimiento y dificultades para la alimentación se confunden con otros síndromes como el síndrome de Bohring-Opitz (BOS, MIM # 605039) (tabla 1)12,13. El BOS se asocia a variantes génicas en ASXL1 y no se han detectado variantes en ASXL3 por lo que se considera que estos síndromes pudieran ser 2 entidades distintas3,12. La mayoría de las variantes descritas en ASXL3 son de novo y producen codones de terminación prematura o cambio del marco de lectura en la región codificante del gen ASXL3 y se agrupan en una región en el exón 113,8,9. El síndrome de Bainbridge-Ropers representa un desafío para el diagnóstico molecular mediante pruebas de un solo gen o paneles multigénicos, por lo cual, la incorporación reciente de técnicas de secuenciación genómica completa o de secuenciación exómica, son útiles para continuar la pesquisa de la etiología de los trastornos del neurodesarrollo.
Comparación de las características clínicas de los pacientes con síndrome de Bainbridge-Ropers con variantes en ASXL3 y los pacientes reportados con BOS
| Fenotipo | Bainbridge et al., 20133; Dinwiddie et al., 20138; Hori et al., 201610; Srivastava et al., 201611 | Reporte actual | BOS Hastings et al., 201112 |
|---|---|---|---|
| Prenatal | |||
| Retraso de crecimiento intrauterino | 5/9 | No | 11/14 |
| Neurológico | |||
| Retraso global del desarrollo | 9/9 | Sí | 14/14 |
| Hipotonía | 8/9 | Sí | 7/14 |
| Dificultad para la alimentación | 8/9 | Sí | 14/14 |
| Retraso de lenguaje | 6/9 | Sí | — |
| Crisis convulsivas | 2/9 | Sí | — |
| Craneofacial | |||
| Microcefalia | 5/9 | Sí | 14/14 |
| Frente prominente | 5/9 | Sí | — |
| Cejas arqueadas | 5/9 | Sí | — |
| hipertelorismo | 1/9 | Sí | 9/14 |
| Narinas antevertidas/hipoplásicas | 6/9 | Sí | 8/14 |
| Orejas de implantación baja/rotación posterior | 6/9 | SÍ | 8/14 |
| Postura BOS | 0/9 | No | 14/14 |
BOS: síndrome de Bohring-Opitz.
Agradecemos a los familiares del paciente por el interés genuino de compartir la información de su hijo con fines de ampliar el conocimiento científico.
Este trabajo se presentó como trabajo libre en modalidad cartel en el XL Congreso Nacional de Genética Humana en Monterrey, Nuevo León, 11-14 de noviembre de 2015.

