




La enfermedad de Creutzfeldt-Jakob (ECJ) es la enfermedad priónica más frecuente en humanos. Se produce por acúmulo de proteína priónica anormalmente plegada en el Sistema Nervioso Central. Etiológicamente se clasifica en esporádica, adquirida o genética1. Un 10-15% de los casos de ECJ se deben a mutaciones del gen de la proteína priónica (PRNP), siendo la E200K la más frecuente. Se presenta como deterioro cognitivo rápidamente progresivo, signos cerebelosos y mioclonías con un curso progresivo fatal. Las crisis epilépticas aparecen en menos del 15% de los casos de formas esporádicas de ECJ2,3.
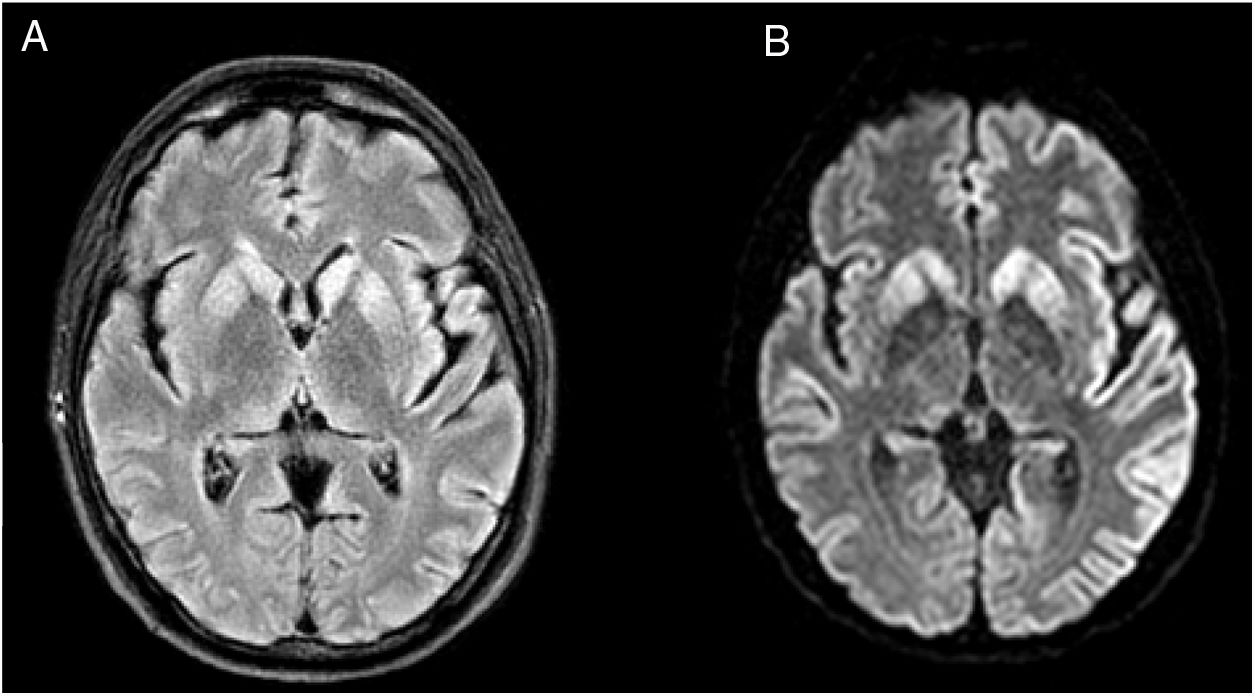
Presentamos el caso de un varón de 48 años de raza caucásica con antecedentes personales de hipertensión arterial, dislipemia y consumo crónico de tabaco y cannabis. Sin antecedentes familiares de interés ni tratamientos previos. Ingresó por alteración de la emisión del lenguaje y de la marcha de dos meses de evolución. A su llegada el paciente presentaba afasia de predominio motor y ataxia de la marcha. En la exploración física destacaba disfasia, conducta desinhibida con agitación psicomotriz y risa inmotivada, grasping bilateral, distonía en mano izquierda, dismetría en miembros superiores, mioclonías multifocales estimulo-inducidas y ataxia cerebelosa de la marcha. Se realizó analítica sanguínea completa incluyendo serologías y autoinmunidad sin alteraciones. El líquido cefalorraquídeo (LCR) fue acelular con leve hiperproteinorraquia, estudio microbiológico negativo y positivo para proteína 14-3-3. En la resonancia magnética cerebral (RM) se observó en secuencia FLAIR, una hiperintensidad de señal en ambos núcleos caudados y una restricción a la difusión en putamen, caudado bilateral así como áreas parasagitales frontal y temporal y giro cingular bilateral con predominio izquierdo (fig. 1). Al tercer día de ingreso presentó episodio de dos a tres minutos de duración de desviación ocular tónica a la derecha y alteración del nivel de conciencia sugestivo de crisis focal secundariamente generalizada. En las dos primeras semanas del ingreso se realizaron hasta seis electroencefalogramas (EEG) donde destacan ondas bi/trifásicas de puntas-polipuntas de forma generalizada con predominio izquierdo que se repiten de forma pseudoperiódica (fig. 2). Pese a tratamiento antiepiléptico con levetiracetam 2.000 mg cada 24 horas, el día 11 de ingreso presentó status epiléptico focal con generalización secundaria con correlato en EEG y respuesta a clonazepam. Precisó ingreso en la Unidad de Cuidados Intensivos (UCI), con mala evolución pese a múltiple tratamiento antiepiléptico (levetiracetam, lacosamida, fenitoína, clonazepam, midazolam y propofol) e inmunomodulador con corticoides e inmunoglobulinas. Finalmente, se confirmó mediante test genético ECJ por mutación E200K del gen PRNP en homocigosis para metionina (MM) en codón 129. El paciente falleció tres semanas después del ingreso. En el estudio neuropatológico se observó vacuolización y astrocitosis transcortical generalizada y particularmente intensa en regiones límbicas y neocorteza temporal compatible con ECJ.

 y EEG de la derecha (5° día de ingreso). Complejos de ondas agudas bi/trifásicas generalizadas de predominio izquierdo que se repiten con una periodicidad variable.")
El cuadro clínico de deterioro cognitivo y ataxia rápidamente progresivo asociando mioclonías multifocales y distonía focal hizo pensar en una encefalopatía priónica como primera posibilidad. El diagnóstico diferencial incluyó encefalitis autoinmunes, encefalitis infecciosas, encefalopatías tóxico-carenciales y metabólicas, vasculitis y carcinomatosis leptomeníngea. La aparición de status epiléptico nos hizo considerar, una vez descartada la encefalitis herpética, la posibilidad de una encefalitis autoinmune e iniciar tratamiento empírico con corticoides e inmunoglobulinas. Además del cuadro clínico característico y de su rápida progresión, una de las claves para sospechar el diagnóstico de ECJ fue la presencia de mioclonías.
La ECJ es una rara enfermedad neurodegenerativa cuyo diagnóstico probable se basa en hallazgos clínicos, electroencefalográficos, neuroimagen y laboratorio. La forma genética tiene una herencia autosómica dominante, pero como ocurre en el caso del paciente, más del 60% no tienen historia familiar previa conocida4,5. Por este motivo, el diagnóstico de un caso genético puede ser difícil.
En fases iniciales de la enfermedad, el EEG puede ser normal, presentar ritmo theta o delta difuso inespecífico, actividades delta rítmicas frontales (FIRDAs), o descargas epileptiformes lateralizadas periódicas (PLEDs). El trazado típico de complejos agudos periódicos (PSWC), suele aparecer en fases avanzadas, en 2/3 de los pacientes con ECJ esporádico y aproximadamente en el 10% de formas genéticas6. Los PSWC se correlacionan con afectación cortical en la RM, más frecuente en formas esporádicas7. El status epiléptico se ha descrito de forma excepcional en la ECJ esporádica8 y sólo dos casos en ECJ familiar9,10.
El paciente presentó una forma genética de la enfermedad sin antecedentes familiares y cursó con un status epiléptico que es infrecuente en formas esporádicas y aún más en formas genéticas. Aunque no presentó en EEG los típicos PSWC, gracias a que se realizaron EEGs seriados pudimos ver complejos bi/trifásicos pseudoperiódicos que, si bien no figuran como criterio diagnóstico, en el contexto clínico de nuestro paciente son sugestivos de ECJ. La mayor afectación cortical temporal observada en neuroimagen y confirmada en estudio neuropatológico pudo ser determinante en la aparición de crisis epilépticas y, por ende, en una evolución más rápida.
En conclusión, este caso ilustra la variabilidad fenotípica de la ECJ, siendo uno de los pocos casos publicados que cursa como status epiléptico focal refractario en una forma genética de la enfermedad. Además, nos permite destacar la importancia de realizar EEGs de forma seriada y estudio genético ante la sospecha de enfermedad priónica.
Comunicación en congresosTrabajo comunicado previamente en la LXXI Reunión Anual de la Sociedad Española de Neurología.
FinanciaciónEste trabajo no ha recibido financiación pública ni privada.
Conflicto de interesesLos autores confirman que no existe ningún conflicto de intereses.

