



Las neuroxinas son una pequeña familia de proteínas de adhesión que participan en la sinapsis mediante la unión al ligando post-sináptico neuroliginas formando complejos trans-sinápticos dependientes de calcio en el sistema nervioso central1–3. En el genoma humano se identifican 3 genes para la transcripción de neuroxinas (NRXN1, NRXN2 y NRXN3). El gen NRXN1 que se encuentra en la región 2p16.3, ocupa una longitud de 1,1Mb e incluye aproximadamente 24 exones4.
Presentamos el caso de un niño en la etapa de la primera infancia, de género masculino, procedente del suroccidente de Colombia. El paciente es producto de la primera gesta entre padres no consanguíneos, con antecedente de atención del parto a término. Durante la valoración inicial del recién nacido se evidencio criptorquidia derecha y se auscultó soplo cardiaco, por lo cual se le realizó ecocardiograma evidenciando estenosis pulmonar periférica, insuficiencia tricuspídea e hipertrofia ventricular izquierda.
El paciente es remitido a los 4 años de edad a la consulta de genética pediátrica, donde al examen físico se encuentra paciente con dolicocefalia, frente prominente, pelo y piel tosca y seca, escases de pelo bitemporal, nariz bulbosa, clinodactilia bilateral en miembros superiores, comportamiento autista (incluyendo auto-agresión) y déficit cognitivo.
Inicialmente al paciente se le realizaron varios estudios que incluían cariotipo bandeo G (46, XY sin anormalidades numéricas o estructurales), escanografía y resonancia magnética que evidencian asimetría del sistema ventricular posterior con dilatación ventricular derecha, cambios anatómicos en el piso ventricular y megacisterna magna, sin otras alteraciones.
Se le realizaron estudios de tamizaje metabólico, hibridación genómica comparativa Array y panel molecular para rasopatías, con resultados negativos. La valoración neuropsicológica informó de la presencia de autismo en la infancia severo con una puntuación del espectro autista de 94 (mínimo 24/máximo 96). Se solicitó panel molecular para autismo, que incluyó 101 genes entre ellos: NRXN1, evidenciando 2 mutaciones en heterocigosis. La primera consistente en un cambio de una citosina por una timina en la posición 1405 (c.1405C>T) que produce, presumiblemente, el cambio de una prolina por una serina en la posición 469 (p. Pro469Ser) [rs7850316]; y la segunda el cambio de una adenina por una guanina en la posición 4053 (c.4053A>G), en donde el cambio es de alanina por Alanina en la posición 1351 (p. Ala1351Ala) [rs7997075]. Los padres fueron valorados para ambas mutaciones a encontrando que el padre es portador heterocigoto de la variante c.1405C>T (p.Pro469Ser) [rs7850316] y la madre es portadora heterocigota de la variante c.4053A>G (p.Ala1351Ala) [rs7997075].
Actualmente en la literatura se identifican 3 casos de pacientes con mutaciones bialélicas del gen NRXN1 y fenotipo similar, a los cuales se les denominó síndrome de Pitt-Hopkins like 25,6. Se considera que este paciente podría ser el cuarto caso de síndrome de Pitt-Hopkins like 2, pero el primero en presentar una mutación heterocigota del gen NRXN1, no asociado a deleciones en la región 2p16.3.
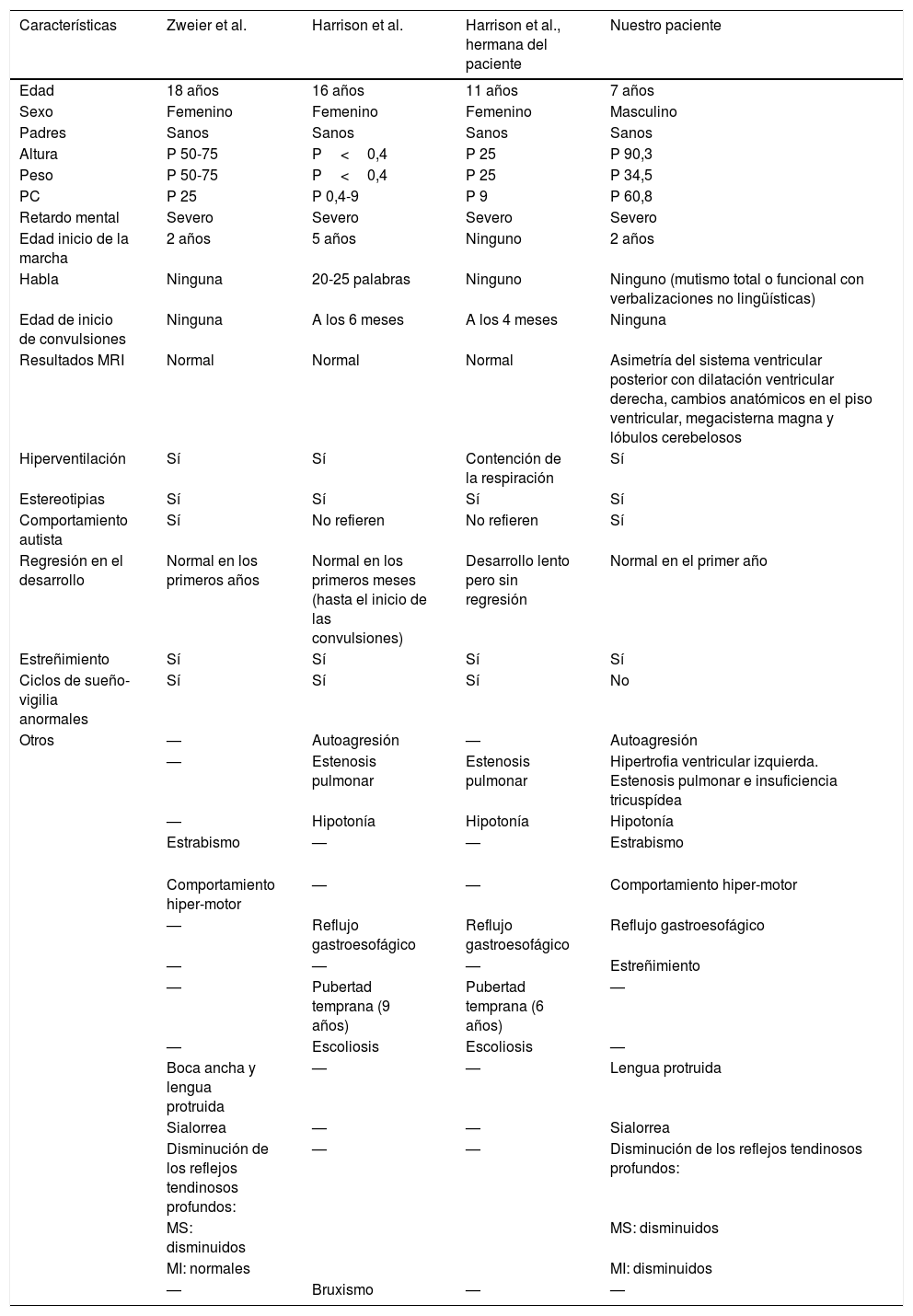
La variante c.1405C>T (rs78540316), es calificada en la base de datos Clinical Variant (ClinVar) como un cambio de significado clínico incierto. Al realizar análisis in silico de esta variante, los resultados son contradictorios, ya que el software SIFT predice que probablemente se trate de un cambio tolerado, mientras el software PolyPhen-2 predice que probablemente se trate de un cambio patológico. Sin embargo, un análisis comparativo entre el fenotipo del caso actual, con los casos descritos por Zweier et al., 2009 y Harris et al., 2011 (tabla 1), muestran una congruencia importante entre los signos y síntomas; excepto por la asimetría del sistema ventricular posterior con dilatación ventricular derecha, los cambios anatómicos en el piso ventricular, y megacisterna magna5–7. Estos hallazgos podrían ser nueva adición al espectro fenotípico del síndrome Pitt-Hopkins like 2, y probablemente se deban a las mutaciones particulares que presenta el paciente reportado.
Comparación fenotipo pacientes reportados en la literatura con del síndrome Pitt-Hopkins like 2 y el caso actual
| Características | Zweier et al. | Harrison et al. | Harrison et al., hermana del paciente | Nuestro paciente |
|---|---|---|---|---|
| Edad | 18 años | 16 años | 11 años | 7 años |
| Sexo | Femenino | Femenino | Femenino | Masculino |
| Padres | Sanos | Sanos | Sanos | Sanos |
| Altura | P 50-75 | P<0,4 | P 25 | P 90,3 |
| Peso | P 50-75 | P<0,4 | P 25 | P 34,5 |
| PC | P 25 | P 0,4-9 | P 9 | P 60,8 |
| Retardo mental | Severo | Severo | Severo | Severo |
| Edad inicio de la marcha | 2 años | 5 años | Ninguno | 2 años |
| Habla | Ninguna | 20-25 palabras | Ninguno | Ninguno (mutismo total o funcional con verbalizaciones no lingüísticas) |
| Edad de inicio de convulsiones | Ninguna | A los 6 meses | A los 4 meses | Ninguna |
| Resultados MRI | Normal | Normal | Normal | Asimetría del sistema ventricular posterior con dilatación ventricular derecha, cambios anatómicos en el piso ventricular, megacisterna magna y lóbulos cerebelosos |
| Hiperventilación | Sí | Sí | Contención de la respiración | Sí |
| Estereotipias | Sí | Sí | Sí | Sí |
| Comportamiento autista | Sí | No refieren | No refieren | Sí |
| Regresión en el desarrollo | Normal en los primeros años | Normal en los primeros meses (hasta el inicio de las convulsiones) | Desarrollo lento pero sin regresión | Normal en el primer año |
| Estreñimiento | Sí | Sí | Sí | Sí |
| Ciclos de sueño-vigilia anormales | Sí | Sí | Sí | No |
| Otros | — | Autoagresión | — | Autoagresión |
| — | Estenosis pulmonar | Estenosis pulmonar | Hipertrofia ventricular izquierda. Estenosis pulmonar e insuficiencia tricuspídea | |
| — | Hipotonía | Hipotonía | Hipotonía | |
| Estrabismo | — | — | Estrabismo | |
| Comportamiento hiper-motor | — | — | Comportamiento hiper-motor | |
| — | Reflujo gastroesofágico | Reflujo gastroesofágico | Reflujo gastroesofágico | |
| — | — | — | Estreñimiento | |
| — | Pubertad temprana (9 años) | Pubertad temprana (6 años) | — | |
| — | Escoliosis | Escoliosis | — | |
| Boca ancha y lengua protruida | — | — | Lengua protruida | |
| Sialorrea | — | — | Sialorrea | |
| Disminución de los reflejos tendinosos profundos: | — | — | Disminución de los reflejos tendinosos profundos: | |
| MS: disminuidos | MS: disminuidos | |||
| MI: normales | MI: disminuidos | |||
| — | Bruxismo | — | — |
Es importante mencionar, que los casos previamente descritos de Pitt-Hopkins like 2, presentan una mutación tipo deleción en homocigosis que afecta el gen NRXN15–7, lo cual hace suponer una herencia de tipo autosómica recesiva; sin embargo el caso actual presenta una mutación heterocigota de tipo de sustitución de nucleótido, también evidenciada en el padre del menor, por lo que se considera que en este caso en particular, la mutación (c.1405C>T) tienen un comportamiento del tipo autosómico dominante con penetrancia incompleta, o es causado por un efecto aditivo entre las mutaciones c.1405C>T y c.4053A>G, haciendo de nuestro paciente un heterocigoto compuesto para NRXN, lo que justificaría el fenotipo sano de los padres.
Se considera que la importante congruencia en el fenotipo de los pacientes previamente reportados y el caso actual, en asociación a la presencia de una mutación en el gen NRXN1, hace pensar que el paciente presenta la misma enfermedad, dando soporte a la noción de que mutaciones en el gen NRXN1 son causales de un fenotipo consistente en autismo y déficit cognitivo similar al síndrome de Pitt-Hopkins.

