







la xantomatosis cerebro-tendinosa (XCT) es una enfermedad autosómica recesiva producida por un déficit de la enzima 27-hidroxilasa. Como consecuencia, existe una deficiencia de ácido quenodeoxicólico y una sobreproducción de colestanol que se deposita en los tejidos. Clínicamente cursa con cataratas, diarrea, xantomas y diferentes síntomas neurológicos. A pesar de que los niveles de colestanol se emplean en el diagnóstico de la XCT, se desconoce su correlación con la clínica y el pronóstico.
Métodosse han revisado 14 pacientes afectos de XCT, diagnosticados entre 1995 y 2008 en dos centros de referencia para el diagnóstico genético, en los que se había determinado el colestanol. Se han estudiado los principales datos demográficos, clínicos y terapéuticos y su posible relación con los niveles de colestanol.
Resultadosla media de los niveles de colestanol al diagnóstico fue de 106μmol/ l. No se encontró ninguna relación entre el colestanol plasmático y los diferentes síntomas neurológicos, ni con el grado de discapacidad al diagnóstico medido mediante la EDSS. Tras la instauración del tratamiento se obtuvo una reducción significativa del colestanol plasmático en todos los casos (reducción media de 91μmol/ l en una media de 34 meses), a pesar de lo cual sólo un paciente se estabilizó clínicamente.
Conclusionesla presencia de niveles elevados de colestanol es muy útil para el diagnóstico de la XCT, pero no tiene valor pronóstico (no se correlaciona con la situación funcional). Su normalización no siempre se acompaña de una estabilización clínica, pero su monitorización puede ser útil para el ajuste del tratamiento.
cerebrotendinous xanthomatosis (CTX) is an autosomal recessive disease caused by a deficiency of mitochondrial enzyme sterol 27-hydrolylase. Such a deficiency results in a reduced production of chenodeoxycholic acid and in an increased formation of cholestanol. It is clinically characterized by cataracts, diarrhoea, xanthomas, premature arteriosclerosis and a number of progressive neurological symptoms. Although cholestanol levels are used for the diagnosis of CTX, their correlation with the clinical symptoms and their prognostic usefulness have not been assessed so far.
Methodswe reviewed 14 CTX patients diagnosed between 1995 and 2008 in two reference centres for the genetic diagnosis of this disorder, whose cholestanol levels had been recorded. We studied the main demographic, clinical and therapeutical data and their correlation with plasma cholestanol levels.
Resultsthe average cholestanol level at diagnosis was 105.8μmol/l. These levels did not correlate with any neurological symptoms or with disability at diagnosis scored by the EDSS. After treatment, all patients achieved a significant reduction in plasma cholestanol levels (average reduction of 91μmol/l in an average follow-up of 34 months), although only one patient remained clinically stable.
Conclusionshigh cholestanol levels are very useful for diagnosis of CTX but they do not have a prognostic value (they do not correlate with severity). Normalisation of cholestanol levels is not always associated with clinical stabilisation. However, follow-up of cholestanol levels can be useful for the dose adjustment.
La xantomatosis cerebrotendinosa (XCT, OMIM # 213700) es una enfermedad autosómica recesiva producida por un déficit en la enzima mitocondrial esterol 27-hidroxilasa (CYP27A1)1 descrita por van Bogaert et al2 en 1937.
Desde el punto de vista clínico los pacientes con XCT suelen presentar una historia de diarrea crónica desde la infancia3. En la tercera década de la vida cursan con cataratas bilaterales y xantomas tendinosos. Posteriormente aparecen los síntomas neurológicos que pueden ser muy variados: ataxia, paraparesia, deterioro cognitivo, trastornos psiquiátricos, parkinsonismo4, polineuropatía5,6, epilepsia y otros1,3-5. Aunque los xantomas tendinosos dan nombre a la enfermedad, su ausencia es frecuente, por lo que en ningún caso excluye el diagnóstico7.
El defecto metabólico primario en la XCT consiste en la alteración de la síntesis de ácidos biliares a partir del colesterol, y es debido a mutaciones en el gen CY27A1, que codifica la enzima mitocondrial esterol 27-hidroxilasa1,8,9. La deficiencia en CYP27A1 conduce especialmente a la reducción de la síntesis de ácido quenodeoxicólico (AQDC) y a un aumento de productos intermediarios, como la 7α-hidroxi-4-colesten-3-ona y de su producto de oxidación, el colestanol. El colestanol también se produce directamente a partir de la oxidación del colesterol y se acumula en todos los tejidos, especialmente en el cerebro, los pulmones, el cristalino y los tendones de los pacientes con XCT, siendo utilizado como marcador bioquímico de esta entidad3. Niveles bajos de colestanol están presentes en la mayoría de los tejidos en mamíferos, y su absorción, como la de los fitoesteroles, es muy reducida en humanos.
No obstante, una elevación del colestanol plasmático puede verse en la cirrosis biliar primaria y en casos de colestasis10,11 como consecuencia de la alteración en la homeostasis del colesterol. Por otra parte, los niveles de colesterol en el plasma y el perfil de lipoproteínas en los pacientes XCT suelen estar normales o por debajo del rango (el perfil de lipoproteínas suele ser “antiaterogénico”)3.
La resonancia magnética (RM) craneal suele mostrar una hiperintensidad al nivel de los núcleos dentados y de los tractos piramidales12. Algunos autores han descrito la potencial utilidad de pruebas funcionales, como el DATSCAN, para evaluar el impacto de esta enfermedad en el sistema nervioso central13.
El tratamiento con AQDC, que inhibe la síntesis anómala de ácidos biliares, es eficaz para corregir las alteraciones bioquímicas14, y según algunos autores enlentece la progresión de la enfermedad3. En el tratamiento se suelen asociar inhibidores de la hidroximetilglutaril-coenzima A (HMG-CoA) reductasa3.
A pesar de que los niveles de colestanol se emplean en el diagnóstico de esta enfermedad desde hace muchos años, su posible valor pronóstico y su utilidad en la monitorización de la evolución de la enfermedad no ha sido muy estudiado. Presentamos un análisis retrospectivo de pacientes españoles con XCT para evaluar la utilidad de los niveles de colestanol en esta enfermedad.
Pacientes y métodosSe revisaron los 26 pacientes con XCT (19 familias) con diagnóstico molecular confirmado en la Fundación Pública Galega de Medicina Xenómica de Santiago de Compostela y en el Hospital Ramón y Cajal de Madrid entre los años 1995 y 2008.
Se registraron los principales datos demográficos (sexo, procedencia, fecha de nacimiento, edad al inicio de los síntomas, edad al diagnóstico, edad al fallecimiento), clínicos (cataratas, xantomas, diarrea, piramidalismo, ataxia, parkinsonismo, retraso psicomotor, deterioro cognitivo, alteraciones psiquiátricas, polineuropatía, epilepsia), pruebas complementarias (TC craneal, RM craneal, RM espinal, estudio genético, estudio neurofisiológico), así como el tratamiento recibido y la evolución. A través de la Expanded Disability Status Scale (EDSS) de Kurtzke15 estimada al diagnóstico se valoró el grado de discapacidad en los pacientes. Puesto que este estudio ha sido realizado por neurólogos, y que con frecuencia los síntomas sistémicos como la diarrea se ven afectados por el sesgo de memoria, se estableció como edad de inicio de la enfermedad aquella en la que se referían los primeros síntomas neurológicos. En los pacientes que comenzaron con síntomas en la infancia (por ejemplo con retraso en la escolarización) y que no recordaban la edad exacta se asignó una edad de inicio de 12 años.
De los 26 pacientes diagnosticados se seleccionaron para el presente estudio solamente los 14 casos (5 varones y 9 mujeres) de 12 familias a los que se les había realizado la determinación de los niveles de colestanol al diagnóstico. De estas 12 familias 4 procedían del área de Galicia, cuatro de Castilla y León, dos de Andalucía, una de Extremadura y una de Castilla La Mancha. En tan solo 5 casos (36%) se identificó la existencia de consanguinidad.
Las determinaciones de colestanol se realizaron en su mayoría en el Instituto de Bioquímica Clínica del Hospital Clínico de Barcelona. El colestanol, conjuntamente con los restantes esteroles de plasma, fue aislado tras hidrólisis alcalina y extracción con disolvente orgánico, y analizado mediante cromatografía de gases capilar en un cromatógrafo Agilent 7890 equipado con detector de llama, como trimetil-silil-derivado (BSTFA 1%/ TMCS en piridina Sigma). La columna utilizada fue una DB1701 (S&W). Se ulilizó epicoprostanol (Sigma) como estándar interno. Los valores de referencia del colestanol (de 2 a 12,6μmol/ l) corresponden a individuos comprendidos entre 1 y 60 años no afectos de alteración en el metabolismo del colesterol, siendo niveles de entre 36 y 102μmol/ l diagnósticos de la XCT. Además, a la mayor parte de los pacientes se les había realizado análisis de colesterol total, fracciones de colesterol y triglicéridos.
Debido al reducido tamaño muestral, se empleó la prueba U de Man-Whitney para comparar los niveles de colestanol entre los diferentes grupos clínicos. Mediante la correlación de Spearman se compararon los niveles de colestanol en función de las diferentes variables cuantitativas. Se consideró como estadísticamente significativa una p < 0,05, utilizando siempre una significación bilateral. Para el análisis estadístico se utilizó el programa SPSS versión 15.0 para Windows (SPSS Inc).
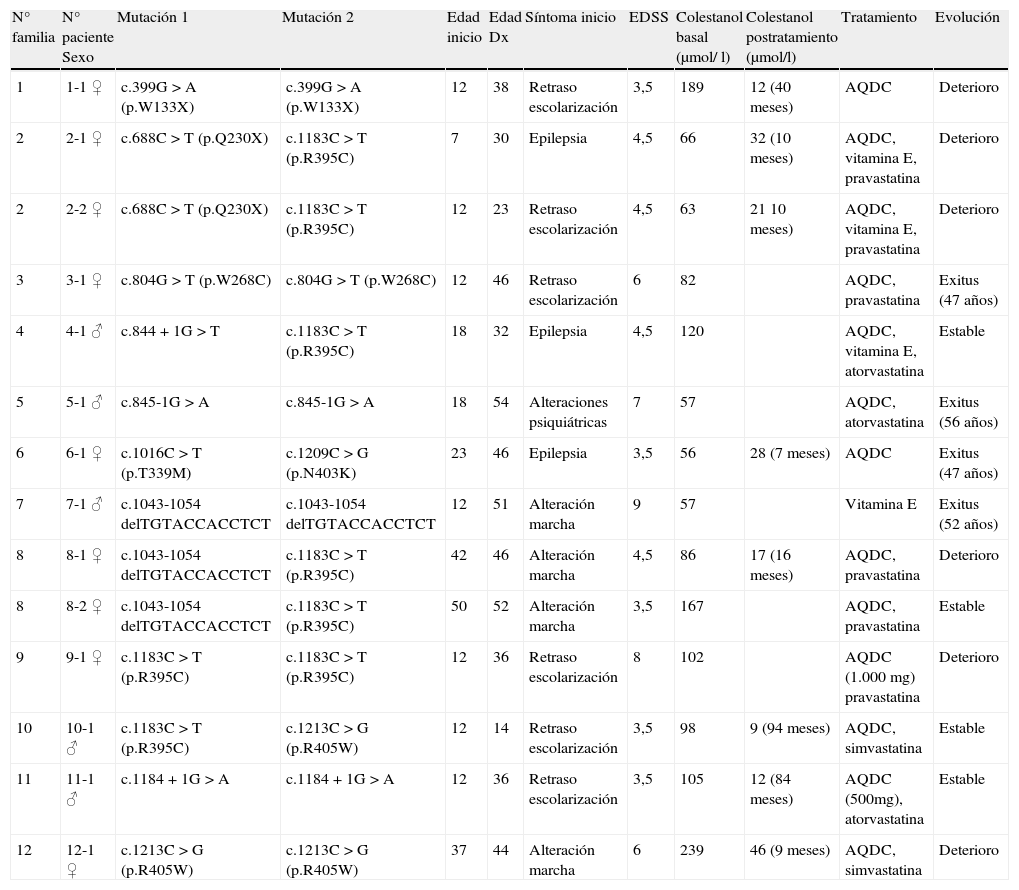
ResultadosEn la tabla 1 se detallan los principales datos demográficos, clínicos, genéticos y bioquímicos de los pacientes incluidos en esta serie, así como su tratamiento y evolución.
Principales datos clínicos, genéticos y bioquímicos de los 14 pacientes con XCT.
| N° familia | N° paciente Sexo | Mutación 1 | Mutación 2 | Edad inicio | Edad Dx | Síntoma inicio | EDSS | Colestanol basal (μmol/ l) | Colestanol postratamiento (μmol/l) | Tratamiento | Evolución |
| 1 | 1-1 ♀ | c.399G>A (p.W133X) | c.399G>A (p.W133X) | 12 | 38 | Retraso escolarización | 3,5 | 189 | 12 (40 meses) | AQDC | Deterioro |
| 2 | 2-1 ♀ | c.688C>T (p.Q230X) | c.1183C>T (p.R395C) | 7 | 30 | Epilepsia | 4,5 | 66 | 32 (10 meses) | AQDC, vitamina E, pravastatina | Deterioro |
| 2 | 2-2 ♀ | c.688C>T (p.Q230X) | c.1183C>T (p.R395C) | 12 | 23 | Retraso escolarización | 4,5 | 63 | 21 10 meses) | AQDC, vitamina E, pravastatina | Deterioro |
| 3 | 3-1 ♀ | c.804G>T (p.W268C) | c.804G>T (p.W268C) | 12 | 46 | Retraso escolarización | 6 | 82 | AQDC, pravastatina | Exitus (47 años) | |
| 4 | 4-1 ♂ | c.844+1G>T | c.1183C>T (p.R395C) | 18 | 32 | Epilepsia | 4,5 | 120 | AQDC, vitamina E, atorvastatina | Estable | |
| 5 | 5-1 ♂ | c.845-1G>A | c.845-1G>A | 18 | 54 | Alteraciones psiquiátricas | 7 | 57 | AQDC, atorvastatina | Exitus (56 años) | |
| 6 | 6-1 ♀ | c.1016C>T (p.T339M) | c.1209C>G (p.N403K) | 23 | 46 | Epilepsia | 3,5 | 56 | 28 (7 meses) | AQDC | Exitus (47 años) |
| 7 | 7-1 ♂ | c.1043-1054 delTGTACCACCTCT | c.1043-1054 delTGTACCACCTCT | 12 | 51 | Alteración marcha | 9 | 57 | Vitamina E | Exitus (52 años) | |
| 8 | 8-1 ♀ | c.1043-1054 delTGTACCACCTCT | c.1183C>T (p.R395C) | 42 | 46 | Alteración marcha | 4,5 | 86 | 17 (16 meses) | AQDC, pravastatina | Deterioro |
| 8 | 8-2 ♀ | c.1043-1054 delTGTACCACCTCT | c.1183C>T (p.R395C) | 50 | 52 | Alteración marcha | 3,5 | 167 | AQDC, pravastatina | Estable | |
| 9 | 9-1 ♀ | c.1183C>T (p.R395C) | c.1183C>T (p.R395C) | 12 | 36 | Retraso escolarización | 8 | 102 | AQDC (1.000 mg) pravastatina | Deterioro | |
| 10 | 10-1 ♂ | c.1183C>T (p.R395C) | c.1213C>G (p.R405W) | 12 | 14 | Retraso escolarización | 3,5 | 98 | 9 (94 meses) | AQDC, simvastatina | Estable |
| 11 | 11-1 ♂ | c.1184+1G>A | c.1184+1G>A | 12 | 36 | Retraso escolarización | 3,5 | 105 | 12 (84 meses) | AQDC (500mg), atorvastatina | Estable |
| 12 | 12-1 ♀ | c.1213C>G (p.R405W) | c.1213C>G (p.R405W) | 37 | 44 | Alteración marcha | 6 | 239 | 46 (9 meses) | AQDC, simvastatina | Deterioro |
En los 14 pacientes con XCT la edad media de inicio de los síntomas neurológicos fue de 20 años (rango de 7 a 50). La edad media al diagnóstico fue de 39 años (rango 14-54), por lo que el retraso medio entre el inicio de los síntomas y su diagnóstico fue de 19 años. Cuatro pacientes habían fallecido en el período comprendido entre su diagnóstico y la realización del estudio.
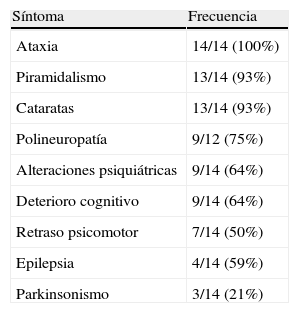
Los síntomas de inicio más frecuentemente encontrados fueron el retraso en la escolarización (43%), la alteración de la marcha (29%) y las crisis epilépticas (21%). La ataxia (100%), el piramidalismo (93%) y las cataratas (93%) fueron los síntomas más frecuentes (tabla 2). Tan sólo el 50% de los pacientes tuvieron xantomas. El EDSS medio calculado al diagnóstico fue de 5,1 (rango: 3,5-9), lo que traduce una discapacidad grave.
Principales síntomas neurológicos encontrados.
| Síntoma | Frecuencia |
| Ataxia | 14/14 (100%) |
| Piramidalismo | 13/14 (93%) |
| Cataratas | 13/14 (93%) |
| Polineuropatía | 9/12 (75%) |
| Alteraciones psiquiátricas | 9/14 (64%) |
| Deterioro cognitivo | 9/14 (64%) |
| Retraso psicomotor | 7/14 (50%) |
| Epilepsia | 4/14 (59%) |
| Parkinsonismo | 3/14 (21%) |
Se identificaron en las 12 familias las dos variantes de secuencia consideradas como las mutaciones patogénicas en cada caso. La mutación más frecuente fue la p.R395C, que se halló hasta en un 25% de los alelos (5 familias).
Los niveles de colestanol medios encontrados fueron de 106μmol/l (rango 56-238) (fig. 1). Se encontró una relación significativa negativa (r=- 0,64; p≤0,013) entre el tiempo de evolución de la enfermedad y los niveles de colestanol, es decir, los niveles de colestanol fueron menores a mayor tiempo de evolución de la enfermedad (fig. 2). No se encontró por el contrario ninguna relación significativa entre el colestanol plasmático y las edades al inicio, al diagnóstico y al fallecimiento. Entre las mujeres se objetivaron unos mayores niveles de colestanol (116μmol/ l, DE 65) que entre los varones (87μmol/ l, DE 28), si bien dado el pequeño tamaño muestral esta diferencia no alcanzó significación estadística. Por otro lado, es de destacar que los niveles de colestanol encontrados en el grupo de pacientes que habían fallecido en el momento del estudio (63μmol/l; rango: 56-82) fueron significativamente inferiores a los obtenidos entre los que seguían vivos en el mismo periodo (123μmol/ l; rango: 63-238) (p ≤ 0,01) (fig. 3A).



Entre los 6 pacientes que tenían xantomas tendinosos se encontraron unos niveles de colestanol más elevados (147μmol/ l; DE: 60), frente a 74μmol/ l (DE: 23) que entre los 8 que no tenían xantomas (fig. 3B) (p=0,01). Tan sólo un paciente de los 14 no tenía cataratas, siendo en este los niveles de colestanol de 57μmol/ l, mientras que entre los otros 11 que sí tenían cataratas la media fue de 109μmol/ l (DE: 56), aunque no se alcanzó significación estadística.
Con respecto a los síntomas neurológicos tan sólo se pudieron relacionar los niveles de colestanol con la presencia de polineuropatía. Entre los 3 pacientes sin neuropatía los niveles hallados fueron de 66μmol/l (DE: 16), mientras que entre los 9 que sí tenían neuropatía fueron de 118μmol/l (DE: 58) (p=0,052). En dos casos, con la revisión de la historia clínica no se pudo confirmar ni descartar la presencia de polineuropatía. Aunque esta diferencia no alcanzó significación estadística, sí puede considerarse una tendencia a la presencia de polineuropatía en los casos con niveles séricos más altos de colestanol. No se encontraron diferencias en los niveles en función del tipo de neuropatía encontrada en el electroneurograma. No se encontró ninguna relación estadísticamente significativa entre los niveles de colestanol y el grado de discapacidad medido a través de la EDSS en el momento del diagnóstico (p=0,3).
Tampoco se encontró ninguna relación estadísticamente significativa entre los niveles de colestanol y los hallazgos radiológicos (tanto en la TC craneal como en la RM craneal y en la RM espinal), con los potenciales evocados ni con el electroencefalograma.
Con respecto al perfil lipídico este fue normal en la mayoría de los casos. Tan sólo en un paciente estaban los niveles de triglicéridos algo elevados (175mg/ dl; N: 40-160mg/ dl), siendo la media de los niveles de triglicéridos de 92mg/ dl. El colesterol total, así como el colesterol LDL encontrados fueron normales en todos los casos, con unas medias respectivas de 174mg/ dl (N: 120-220mg/ dl) y 92,6mg/ dl (N: 60-190mg/ dl). Un paciente presentaba unos niveles de colesterol HDL bajos (24mg/ dl; N: 30-100mg/ dl), siendo la media de los niveles de colesterol HDL de 59mg/ dl. No se encontró ninguna correlación entre los niveles de colestanol y los niveles de colesterol, sus fracciones y triglicéridos.
De los 14 pacientes uno (7,1%) recibió exclusivamente vitamina E (diagnóstico muy tardío; falleció al poco del diagnóstico), dos (14,3%) AQDC exclusivamente, 8 (57,17%) la asociación de AQDC y una estatina y tres (21,4%) la asociación de AQDC, una estatina y vitamina E. Todos los pacientes en tratamiento con AQDC recibieron la dosis estándar (750mg), excepto un paciente que recibió 500mg (paciente 11-1) y otro que recibió 1.000mg (paciente 9-1).
En 8 de los pacientes (57%) se realizó al menos una segunda determinación de los niveles de colestanol tras la instauración del tratamiento para vigilar la evolución de los mismos, objetivándose una reducción significativa en todos los casos e incluso llegándose a normalizar en tres de ellos (37,5%). La reducción media objetivada fue de 91μmol/ l (rango 29-193) en un tiempo medio de 34 meses (rango 7-94) (fig. 4). A pesar de la reducción significativa de los niveles de colestanol en los 8 pacientes monitorizados, sólo uno de ellos (12,5%) se estabilizó desde el punto de vista clínico.

De forma global, a pesar del tratamiento, tan sólo se estabilizaron 4 pacientes de los 14 de la serie (28,6%). Además, 4 pacientes (28,6%) habían fallecido en el periodo comprendido entre el diagnóstico y el presente estudio retrospectivo, con una edad media al fallecimiento de 51 años (rango 47-56).
DiscusiónEl objetivo del presente estudio fue analizar la posible relación entre los niveles plasmáticos de colestanol y diversos parámetros clínicos y pronósticos en la XCT. Para ello, llevamos a cabo una investigación retrospectiva a través de la revisión de las historias clínicas de 14 pacientes españoles con diagnóstico genético y bioquímico de la enfermedad.
El perfil lipídico fue normal en la mayoría de los pacientes. Esto coincide con lo anteriormente publicado en la literatura, que sugiere que los niveles de colesterol y sus fracciones suelen ser normales o algo bajos en los pacientes con XCT16,3.
Los niveles de colestanol medios encontrados fueron de 106μmol/ l, por lo que en todos los pacientes estudiados con XCT los niveles de colestanol estaban muy elevados (entre 5 y 20 veces el valor superior de la normalidad). Cifras elevadas de colestanol no son exclusivas de la XCT y pueden observarse, por ejemplo, en enfermedades hepáticas, fitosterolemia, hipotiroidismo e hiperlipoproteinemia familiar tipo II11,17. También pueden encontrarse elevaciones moderadas en portadores heterozigotos asintomáticos, aunque nunca alcanzan los niveles mínimos detectados en los pacientes con XCT18,19.
Se encontraron unos mayores niveles de colestanol entre aquellos pacientes que tenían xantomas, cataratas y polineuropatía. Estos hallazgos sugieren un depósito de colestanol a nivel de los tendones, de los cristalinos y del sistema nervioso periférico relacionado con mayores niveles circulantes de colestanol, es decir, que en los pacientes con niveles muy elevados estaría favorecido el depósito tisular de colestanol.
Es llamativo el hecho de que no se observara ninguna relación significativa entre los niveles de colestanol y el grado de discapacidad medido a través de la escala EDSS en el momento del diagnóstico. A pesar de que con frecuencia se sobreentienda que unos niveles de colestanol más elevados se relacionan con un peor pronóstico funcional, los resultados de nuestra serie no apoyan dicha interpretación. No obstante, la escala empleada presenta importantes limitaciones al enfatizar en exceso los síntomas motores frente al resto de síntomas no motores y, por lo tanto, infravalorar la complejidad de la enfermedad.
Algunos autores han empleado la monitorización de los niveles de colestanol para el seguimiento terapéutico de los pacientes20. En los 8 pacientes de nuestra serie en los que se realizó al menos una segunda determinación tras la instauración del tratamiento, la reducción fue significativa, llegándose incluso a normalizar los niveles en tres de ellos. Es llamativo, por lo tanto, cómo a pesar de presentar unos niveles muy elevados es posible con la misma dosis de AQDC (la dosis estándar es 750mg) lograr una reducción significativa. Es cuestionable plantear un aumento de la dosis de AQDC en aquellos casos en los que no se normalicen los niveles: la mejoría bioquímica no se correlacionó con una mejoría sintomática, puesto que tan sólo un paciente se logró estabilizar desde el punto de vista clínico. Este paciente había sido diagnosticado a los 14 años y actualmente tiene 22 años, por lo que el principal factor determinante de la estabilización podría haber sido la instauración precoz del tratamiento.
Se encontraron unos niveles de colestanol significativamente inferiores en el grupo de pacientes que fallecieron en relación con los que siguen vivos. Teniendo en cuenta que los pacientes que han fallecido son los que se han diagnosticado de manera más tardía, esto podría indicar que tal vez en las fases más avanzadas de la enfermedad se produzca un descenso en los niveles de colestanol. Esto concuerda con la correlación negativa hallada entre el tiempo de evolución de la enfermedad y los niveles plasmáticos: los pacientes con mayor tiempo de evolución de la enfermedad presentaban unos menores niveles de colestanol.
Otra posible explicación podría ser que los pacientes más graves son aquellos en los que el colestanol se deposita en mayor cantidad en los diferentes tejidos (cristalino, tendones, cerebro) y que por ello es menor en ellos el colestanol circulante en sangre. Nuestros datos ponen también de manifiesto una mala correlación entre los niveles plasmáticos de colestanol y el pronóstico funcional. Se desconoce el motivo por el que el colestanol se deposita selectivamente en unos tejidos (en el tejido nervioso por ejemplo y más concretamente en los núcleos dentados) y por qué algunos pacientes tienen una mayor susceptibilidad a esta acumulación en los tejidos, siendo esta la causante de las lesiones cerebrales irreversibles (pérdida neuronal, cúmulo de cristales lipídicos) descritas en diversos estudios necrópsicos12,21,22.
Por lo tanto, hay que tener precaución a la hora de interpretar el significado de los niveles plasmáticos de colestanol en los pacientes con XCT. Unos niveles elevados son muy útiles para establecer el diagnóstico, pero no tienen valor pronóstico —mayores niveles no se relacionan necesariamente con una peor situación funcional—, ni permiten monitorizar la efectividad clínica del tratamiento —la normalización de los niveles no siempre se acompaña de una estabilización clínica—. Sí pueden por el contrario ser útiles en el ajuste de la dosis de AQDC, así como en la verificación del cumplimiento terapéutico.
La valoración de nuestros resultados presenta varias limitaciones. En primer lugar, existe una importante dispersión de datos, pues a cada paciente se le realizó la determinación de niveles de colestanol en el momento del diagnóstico, que en cada caso se corresponde con un momento diferente de la evolución natural de la enfermedad y a una edad diferente. En segundo lugar desconocemos cuál es la curva de la evolución natural de dichos niveles a lo largo de la enfermedad. Aunque nuestros hallazgos sugieren un incremento inicial en función de la edad de inicio de los síntomas, y posteriormente un descenso paulatino a medida que evoluciona la enfermedad, el diseño del estudio —de tipo transversal — no permite confirmar esta hipótesis. Finalmente, el reducido tamaño muestral (debido a la baja prevalencia de la enfermedad) limita el análisis estadístico de los datos. Son necesarios pues estudios adicionales, con series más extensas y un seguimiento detallado, prospectivo, para documentar con más precisión el valor clínico de los niveles de colestanol en el pronóstico y seguimiento de los pacientes con XCT.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los neurólogos que han colaborado en la realización de este trabajo: Dr. M. Arias (CHUS), Dr. A. Ares (Hospital de León), Dr. J. Duarte (Hospital General de Segovia), Dra. I. García Castañón (Hospital San Pedro Alcántara), Dr. A. López de Munain (Hospital de San Sebastián), Dr. A. Molina (Hospital 12 de Octubre), Dr. M. Moya (Hospital Línea de la Concepción), Dr. D. Ezpeleta (Hospital Gregorio Marañón) y Dr. F.J. Jiménez Jiménez (Hospital del Sureste).
Este trabajo ha sido parcialmente presentado en el 13th Meeting of the European Federation of the Neurological Societies (Florencia, 12-15 de septiembre de 2009) en el póster “Cerebrotendinous Xanthomatosis in Spain”. Este trabajo no ha recibido ningún tipo de financiación.

