



El ganglioglioma es un tumor primario, de bajo grado, del sistema nervioso central constituido por una población celular mixta de elementos gliales y neuronales. Representan entre el 0,4 al 2% de todos los tumores intracraneales y afectan fundamentalmente a niños y adultos jóvenes.
MétodosEntre los años 1995 y 2008 hemos tratado en nuestro hospital 20 pacientes (12 adultos y 8 niños) con ganglioglioma intracraneal. Revisamos retrospectivamente el sexo, el síntoma de inicio y la edad, sintomatología y tiempo de evolución, exploración neurológica, localización del tumor, aspecto en la tomografía computarizada y resonancia magnética, el tratamiento quirúrgico y la evolución. Todos los pacientes fueron intervenidos quirúrgicamente y la extensión de la resección fue evaluada de la hoja operatoria y del seguimiento neurorradiológico.
ResultadosLa media de edad de los pacientes fue de 26,4 años (rango 1-75) y el ratio mujer/varón fue de 1.5:1. Excepto en un caso, todos los pacientes debutaron con crisis epilépticas, con una duración media antes del diagnóstico de 7,4 años (rango 1-29). Diecisiete tumores estaban localizados en el lóbulo temporal (9 derechos y 8 izquierdos). Se realizó extirpación macroscópicamente completa en 17 pacientes y subtotal en los 3 restantes. Se presentaron 4 recidivas que fueron tratadas mediante reintervención, añadiéndose radioterapia en uno de los casos. El tiempo medio de seguimiento fue de 8,5 años (rango 22 meses-14 años), la supervivencia libre de enfermedad a los 5 años fue del 85% y la supervivencia global del 95%.
ConclusionesLas crisis epilépticas, que constituyen el síntoma más frecuente mejoran de forma significativa tras la extirpación quirúrgica. El tratamiento quirúrgico es la primera opción terapéutica en este tipo de tumores, y ante la presencia de resecciones subtotales o recidivas tumorales la mejor indicación de tratamiento es la reintervención. La radioterapia debe reservarse únicamente para las formas malignas.
A ganglioglioma is a type of primary central nervous system low grade tumour composed of mixed populations of glial and neuroepithelial elements. They accounts for 0.4 to 2% of all intracranial tumours and appear more commonly in children and young adults. Seizures, which are the most important symptom in these tumours, improve significantly after surgical excision.
MethodsBetween 1995 and 2008, 20 patients with (12 adults and 8 children) with intracranial ganglioglioma were treated at our hospital. Clinical information obtained by chart review included sex, age at onset of symptoms, clinical history, results of neurological examination, tumour location, CT and MRI appearance, surgical results and follow-up. All patients underwent tumour resection and the extent of surgery was determined from the surgical reports and postoperative imaging studies.
ResultsThe median age of patients was 26.4 years (range, 1-75 years), and the female to male ratio was 1.5:1. Except in one case, all patients had seizures with a median duration before diagnosis of 7.4 years (range 1-29). Seventeen tumours were located in the temporal lobe (9 right and 8 left). Macroscopically complete excision was performed in 17 patients and subtotal in the remaining 3. There were 4 cases of recurrence treated by surgery and radiotherapy being added in one case. The mean follow up was 8.5 years (range 22 months-14 years) and disease free survival at 5 years was 85% and an overall survival of 95%.
ConclusionsThe seizures, which are the most frequent symptoms, significantly improved after surgical removal. Surgery is the first choice of therapy in these tumours, and in the presence of subtotal resection or tumour recurrence the best indication for treatment is repeat surgery. Radiotherapy should be reserved only for malignant forms.
Las neoplasias del sistema nervioso central (SNC) son un grupo muy heterogéneo de tumores con distintas histologías, comportamientos, localizaciones, precisando, por lo tanto, diferentes y complejos enfoques terapéuticos. En la mayoría de casos se trata de tumores de naturaleza glial tanto de bajo como de alto grado o tumores de origen meníngeo. En la clasificación de la Organización Mundial de la Salud (OMS) se incluye un grupo tumoral denominado tumores glioneurales o neurogliales, que representa entre el 0,4-2% de todos los tumores del SNC, en los que se agrupan los tumores caracterizados por la presencia de células ganglionares y elementos gliales (astrocitarios u oligodendrogliales) y que se corresponden habitualmente con grados I y II de la OMS1.
Estos tumores son característicos de la edad pediátrica y adultos jóvenes, tienen preferencia por el lóbulo temporal y cursan en un elevado número de casos con crisis epilépticas de larga evolución2,3.
Presentamos la experiencia de nuestro servicio con 20 casos de gangliogliomas (GG) de hemisferios cerebrales tratados quirúrgicamente en un período de 14 años.
Pacientes y métodoRealizamos un estudio retrospectivo descriptivo sobre 20 pacientes afectos de ganglioglioma supratentorial, diagnosticados y tratados quirúrgicamente en nuestro servicio de Neurocirugía entre los años 1995-2008, correspondiendo 11 casos a pacientes adultos y 9 a pediátricos (menores de 16 años).
Analizamos, además de los datos epidemiológicos básicos, el síntoma de inicio, tiempo de evolución desde el comienzo de la sintomatología hasta el diagnóstico definitivo, los datos de exploración clínica más relevantes, las pruebas de diagnóstico neurofisiológico y neurorradiológicas practicadas, el tratamiento realizado y sus complicaciones, los resultados quirúrgicos y la evolución.
Se ha podido realizar el seguimiento clínico y radiológico en todos los pacientes.
Todas las piezas extirpadas fueron analizadas histológicamente mediante hematoxilina-eosina, empleando las tinciones inmunohistoquímicas específicas.
ResultadosLos 20 tumores analizados constituyen el 1,8% de todos los procesos neoplásicos intervenidos en el servicio en el período de tiempo de 14 años.
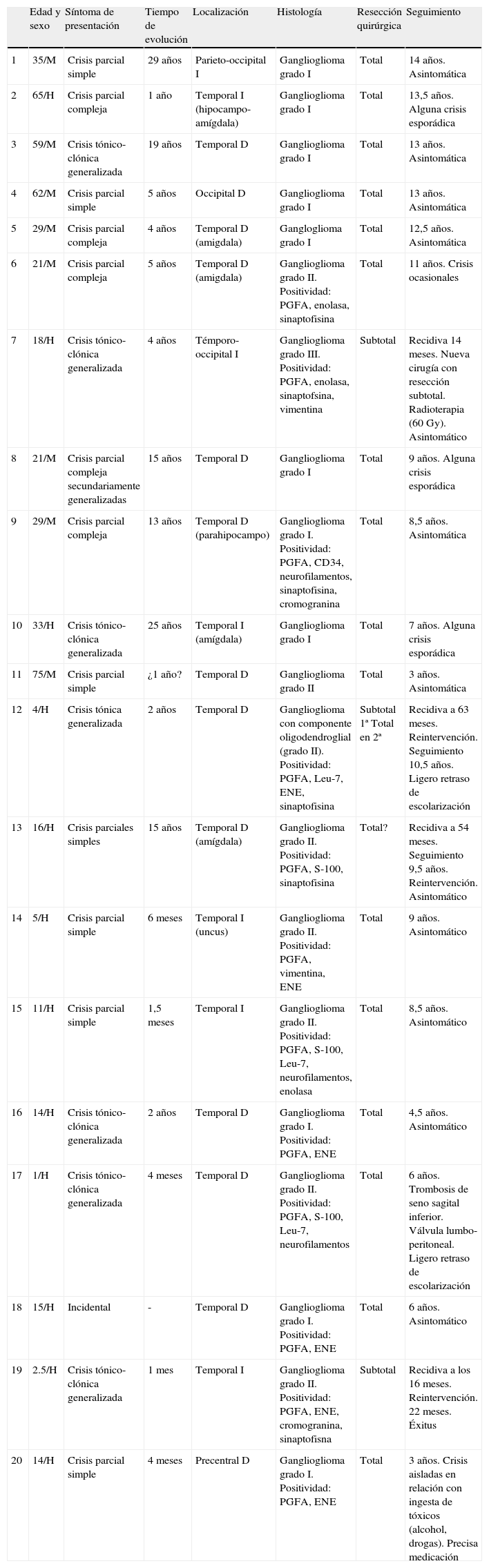
Edad y sexoLa serie consta de 12 varones y 8 mujeres, con edades comprendidas entre 1 y 75 años (media 26,4) (tabla 1).
Datos clínicos y neurofisiológicos de la serie
| Edad y sexo | Síntoma de presentación | Tiempo de evolución | Localización | Histología | Resección quirúrgica | Seguimiento | |
| 1 | 35/M | Crisis parcial simple | 29 años | Parieto-occipital I | Ganglioglioma grado I | Total | 14 años. Asintomática |
| 2 | 65/H | Crisis parcial compleja | 1 año | Temporal I (hipocampo-amígdala) | Ganglioglioma grado I | Total | 13,5 años. Alguna crisis esporádica |
| 3 | 59/M | Crisis tónico-clónica generalizada | 19 años | Temporal D | Ganglioglioma grado I | Total | 13 años. Asintomática |
| 4 | 62/M | Crisis parcial simple | 5 años | Occipital D | Ganglioglioma grado I | Total | 13 años. Asintomática |
| 5 | 29/M | Crisis parcial compleja | 4 años | Temporal D (amigdala) | Gangloglioma grado I | Total | 12,5 años. Asintomática |
| 6 | 21/M | Crisis parcial compleja | 5 años | Temporal D (amigdala) | Ganglioglioma grado II. Positividad: PGFA, enolasa, sinaptofisina | Total | 11 años. Crisis ocasionales |
| 7 | 18/H | Crisis tónico-clónica generalizada | 4 años | Témporo-occipital I | Ganglioglioma grado III. Positividad: PGFA, enolasa, sinaptofsina, vimentina | Subtotal | Recidiva 14 meses. Nueva cirugía con resección subtotal. Radioterapia (60 Gy). Asintomático |
| 8 | 21/M | Crisis parcial compleja secundariamente generalizadas | 15 años | Temporal D | Ganglioglioma grado I | Total | 9 años. Alguna crisis esporádica |
| 9 | 29/M | Crisis parcial compleja | 13 años | Temporal D (parahipocampo) | Ganglioglioma grado I. Positividad: PGFA, CD34, neurofilamentos, sinaptofisina, cromogranina | Total | 8,5 años. Asintomática |
| 10 | 33/H | Crisis tónico-clónica generalizada | 25 años | Temporal I (amígdala) | Ganglioglioma grado I | Total | 7 años. Alguna crisis esporádica |
| 11 | 75/M | Crisis parcial simple | ¿1 año? | Temporal D | Ganglioglioma grado II | Total | 3 años. Asintomática |
| 12 | 4/H | Crisis tónica generalizada | 2 años | Temporal D | Ganglioglioma con componente oligodendroglial (grado II). Positividad: PGFA, Leu-7, ENE, sinaptofisina | Subtotal 1ª Total en 2ª | Recidiva a 63 meses. Reintervención. Seguimiento 10,5 años. Ligero retraso de escolarización |
| 13 | 16/H | Crisis parciales simples | 15 años | Temporal D (amígdala) | Ganglioglioma grado II. Positividad: PGFA, S-100, sinaptofisina | Total? | Recidiva a 54 meses. Seguimiento 9,5 años. Reintervención. Asintomático |
| 14 | 5/H | Crisis parcial simple | 6 meses | Temporal I (uncus) | Ganglioglioma grado II. Positividad: PGFA, vimentina, ENE | Total | 9 años. Asintomático |
| 15 | 11/H | Crisis parcial simple | 1,5 meses | Temporal I | Ganglioglioma grado II. Positividad: PGFA, S-100, Leu-7, neurofilamentos, enolasa | Total | 8,5 años. Asintomático |
| 16 | 14/H | Crisis tónico-clónica generalizada | 2 años | Temporal D | Ganglioglioma grado I. Positividad: PGFA, ENE | Total | 4,5 años. Asintomático |
| 17 | 1/H | Crisis tónico-clónica generalizada | 4 meses | Temporal D | Ganglioglioma grado II. Positividad: PGFA, S-100, Leu-7, neurofilamentos | Total | 6 años. Trombosis de seno sagital inferior. Válvula lumbo-peritoneal. Ligero retraso de escolarización |
| 18 | 15/H | Incidental | - | Temporal D | Ganglioglioma grado I. Positividad: PGFA, ENE | Total | 6 años. Asintomático |
| 19 | 2.5/H | Crisis tónico-clónica generalizada | 1 mes | Temporal I | Ganglioglioma grado II. Positividad: PGFA, ENE, cromogranina, sinaptofisna | Subtotal | Recidiva a los 16 meses. Reintervención. 22 meses. Éxitus |
| 20 | 14/H | Crisis parcial simple | 4 meses | Precentral D | Ganglioglioma grado I. Positividad: PGFA, ENE | Total | 3 años. Crisis aisladas en relación con ingesta de tóxicos (alcohol, drogas). Precisa medicación |
D: derecho; ENE: enolasa neuronal específica; H: hombre; I: izquierdo; M: mujer; PGFA: proteína gliofibrilar ácida.
Exceptuando un paciente de 15 años cuyo diagnóstico fue incidental tras un accidente de circulación, la totalidad de los pacientes debutaron con crisis epilépticas que en 7 casos fueron motoras generalizadas, 7 parciales simples y 5 parciales complejas, con un tiempo de evolución desde el comienzo de los síntomas hasta el diagnóstico definitivo que osciló entre 1 mes y 29 años, siendo en la población infantil de 1-180 meses (media 25 meses) y de 1-29 años en los adultos (media: 10,3 años).
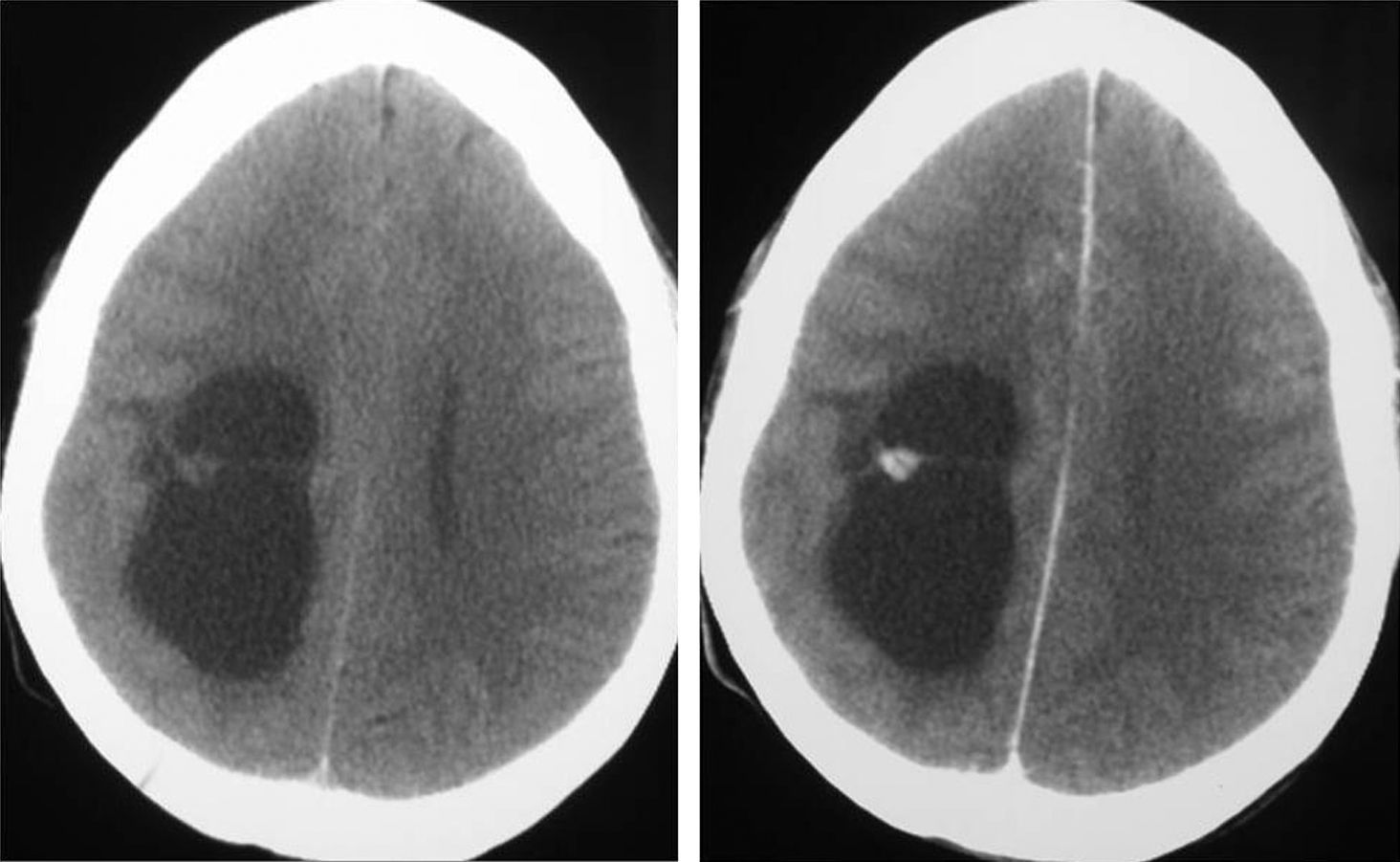
Diagnóstico neurorradiológicoSe realizó estudio con tomografía computarizada (TC) en los 20 pacientes presentándose el tumor como hipodenso en 16 casos, hiperdenso en 2 y heterogéneo en otro; el paciente número 20 presentaba un estudio que fue considerado normal. En 14 casos existía un realce entre ligero y moderado con el contraste y en 5 pacientes no se objetivaba captación (fig. 1).
.")
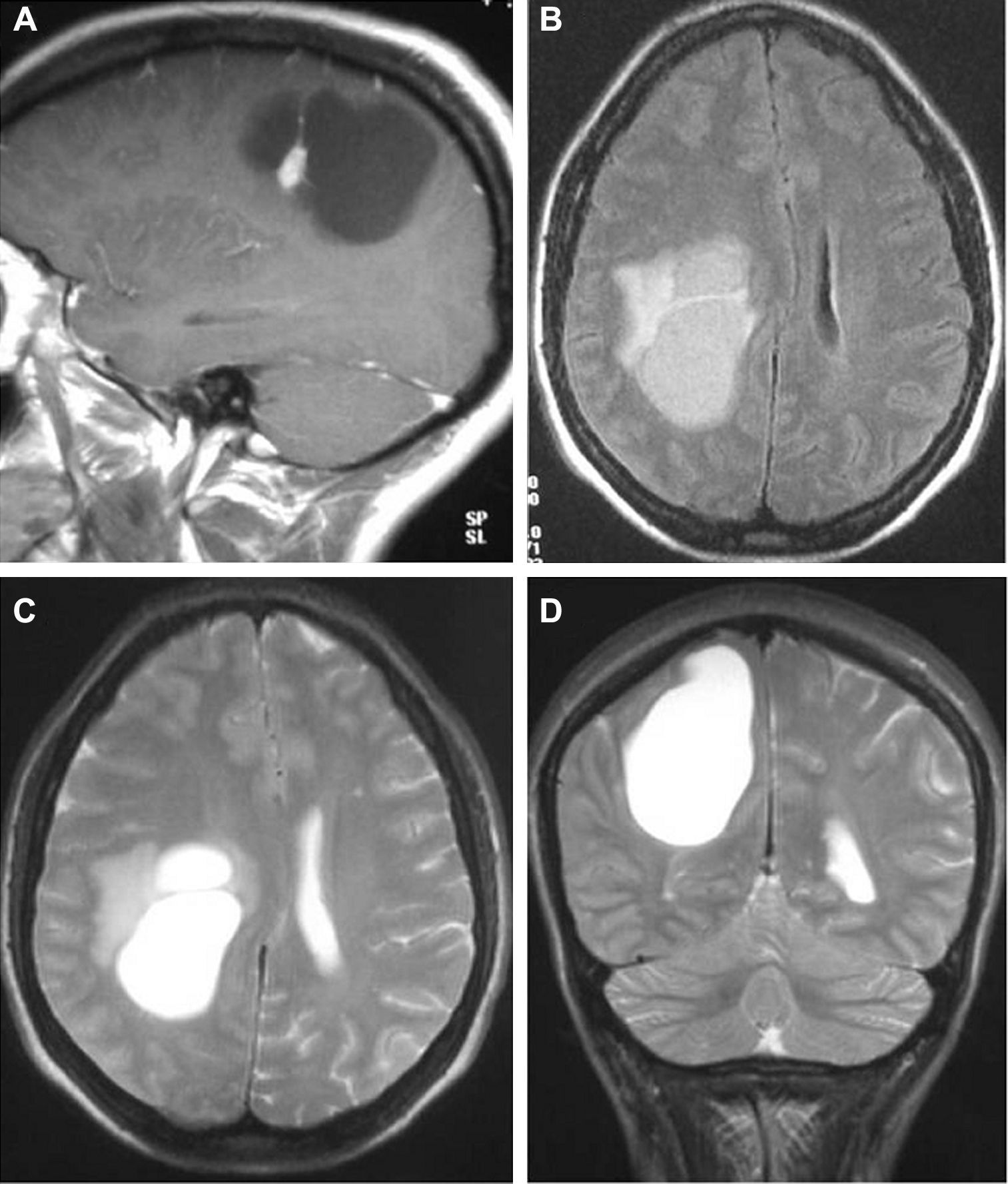
La resonancia magnética (RM), practicada en todos los pacientes, demostró en las secuencias T1 que 12 tumores se presentaban como hipointensos, 6 como hiperintensos, 1 isointenso y el restante de manera heterogénea. En las secuencias T2 todos los gangliogliomas se mostraban como hiperintensos (fig. 2). En todos los casos existía realce con gadolinio entre moderado e intenso. Doce tumores eran quísticos y ocho sólidos.
 Secuencia T1 con contraste. B-D) Secuencias T2.")
Únicamente se realizó estudio angiográfico en un paciente de 2 años (caso número 19) con un voluminoso tumor de localización temporal izquierda observándose un desplazamiento de estructuras vasculares (cerebral media) y una tinción tumoral difusa.
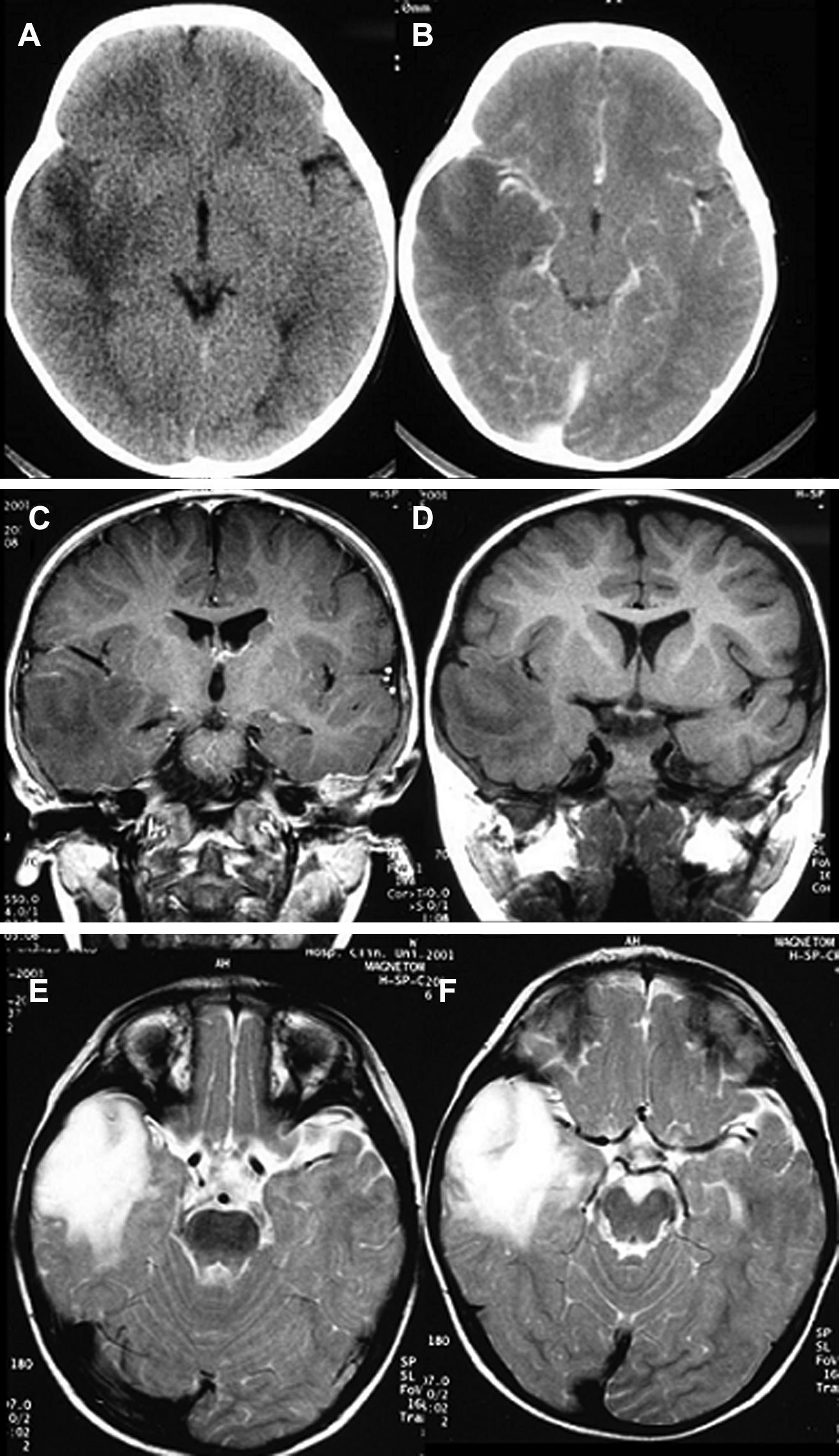
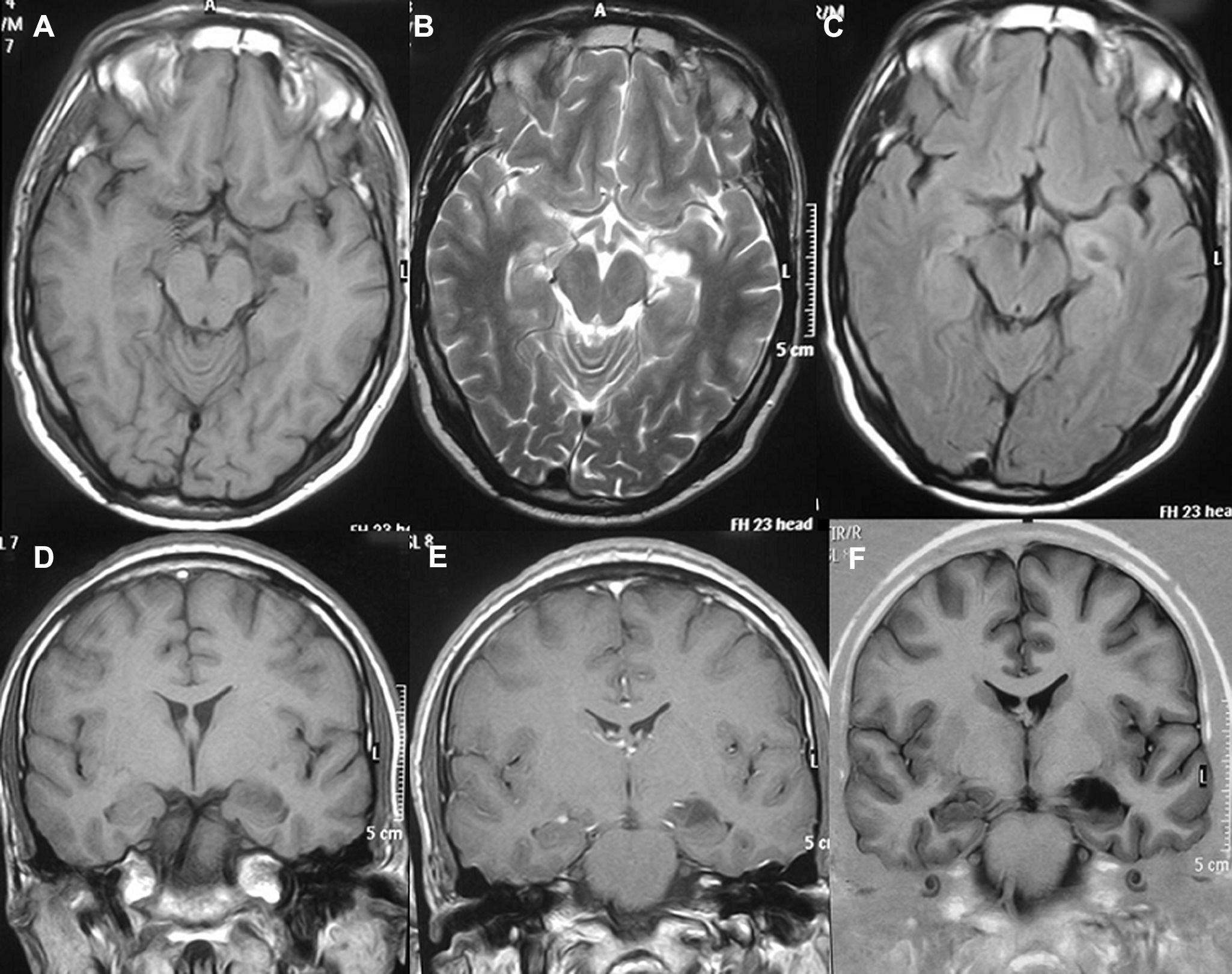
LocalizaciónEn el lóbulo temporal se situaban 17 tumores (9 derechos/8 izquierdos) (figs. 3 y 4) y los otros tres estaban situados en lóbulo occipital (2 casos) y el tercero de localización precentral.
 TAC sin y con contraste correspondiente a un ganglioglioma temporal. C-D) RM secuencias T1 sin y con contraste. E-F: RM secuencias T2 (caso 17).")
, T2 (B) y FLAIR (C), T1 sin y con contraste (D, E) y ecogradiente (caso 10).")
Se practicó estudio electroencefalográfico (EEG) en todos los pacientes, encontrándose diversas formas de alteración en todos los casos sintomáticos, el niño cuyo tumor fue un hallazgo incidental el EEG fue normal. En 6 pacientes se realizó estudio de video-EEG pudiendo evaluar las crisis en 4 casos.
TratamientoTodos los pacientes fueron intervenidos quirúrgicamente, empleando técnica microquirúrgica, se realizo exéresis macroscópicamente total en 17 casos y superior al 75% en los otros 3. En el paciente número 7 en el que realizó una extirpación incompleta se presentó recrecimiento a los 14 meses y tras una segunda cirugía, también incompleta, se realizó radioterapia.
ComplicacionesÚnicamente se presentaron dos complicaciones dignas de mención, el paciente con el ganglioglioma grado III presentó una dilatación ventricular que se solucionó con la implantación de una válvula ventrículo-peritoneal y un niño de 2 años, que a los 3 meses de la cirugía desarrolló un quiste a tensión sobre el lecho operatorio y que obligó a la implantación de una derivación cisto-peritoneal. Meses más tarde, desarrolló un cuadro de hipertensión intracraneal por trombosis del seno sagital inferior, siendo necesaria la implantación de una derivación lumbo-peritoneal retirándose previamente la anterior válvula.
EvoluciónEl tiempo medio de seguimiento fue de 8,5 años (rango 22 meses-14 años). La supervivencia libre de enfermedad a los 5 años para los pacientes con este seguimiento mínimo fue del 85% y la supervivencia global del 95%. De los 18 pacientes que debutaron con epilepsia y que seguían vivos después de 2 años, 5 casos (27,7%) no precisaron medicación anticomicial, siendo esta reducción más significativa en la población pediátrica 3 casos (33,3%) que en los adultos, 2 casos (18,8%).
Fueron reintervenidos 4 pacientes, en tres casos se trataba de recrecimiento de restos tumorales tras exéresis incompletas y el otro caso presentó una recidiva a los 54 meses de haber realizado una extirpación que fue considerada quirúrgica y radiográficamente como completa, en estos casos se trataba de 2 gangliogliomas grado II, un ganglioglioma grado III y otro con componente oligodendroglial. Sólo se presentó un episodio de mortalidad (caso número19) debido a la progresión del tumor.
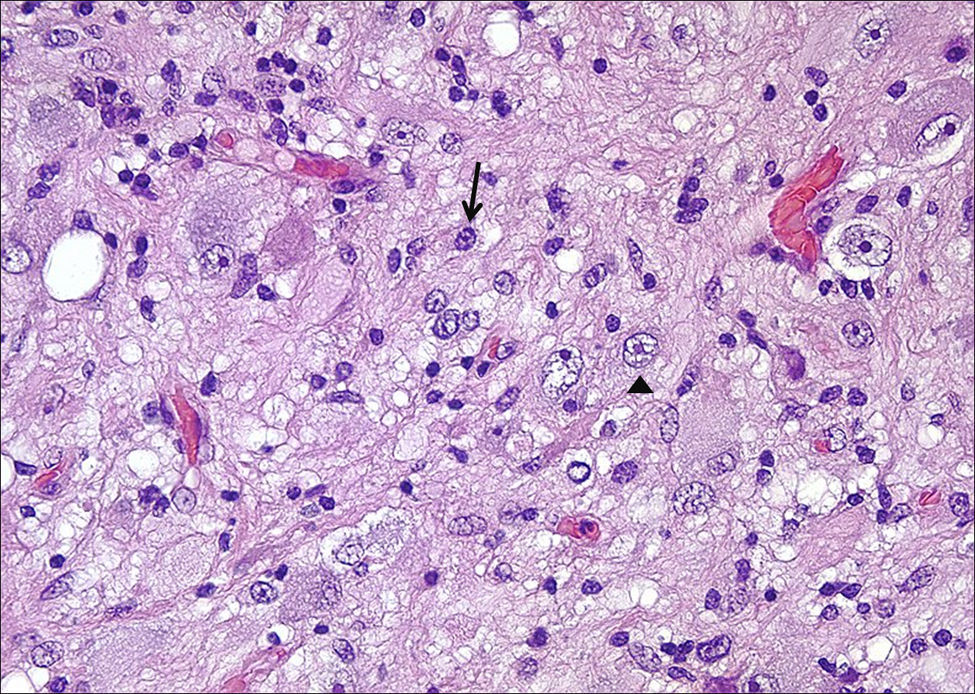
HistologíaTodos las piezas extirpadas fueron estudiadas histológicamente realizándose estudios en hematoxilina-eosina y técnicas de inmuno histoquímica específicas. En 11 casos se trataba de un ganglioglioma típico grado I y en 6 casos grado II de la OMS, otro caso tenía un componente mixto oligodendroglial (grado II) y un tercero se trataba de un ganglioglioma anaplásico grado III de la OMS (figs. 5 y 6). En los 4 casos sometidos a nueva intervención quirúrgica no se observó modificación en el tipo o grado de tumor.
 y neuronal (flecha) (HE 100x).")
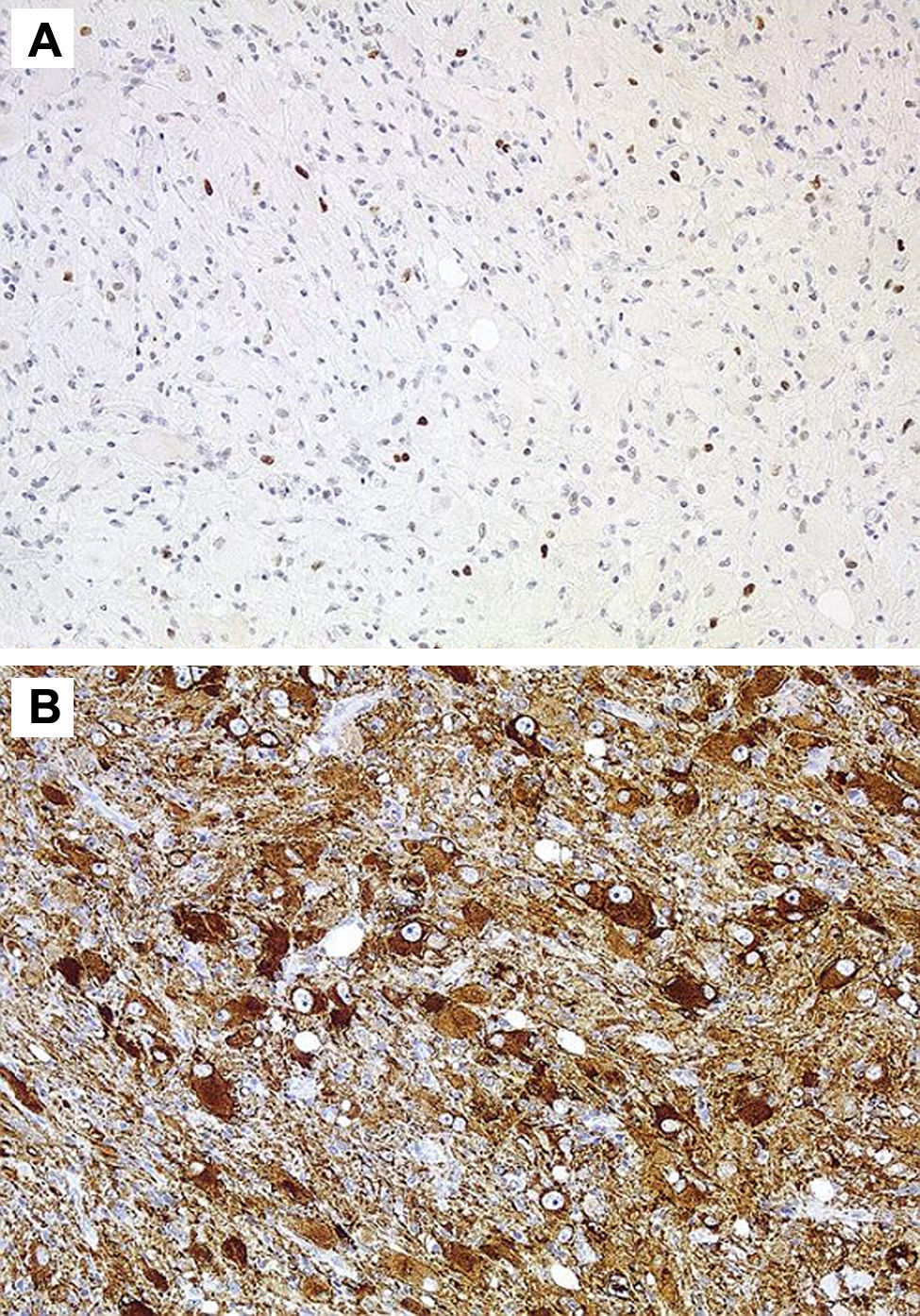
 Positividad muy escasa para P53. B) Positividad para sinaptofisina en las células.")
En 4 casos de ganglioglioma grado II se realizó determinación de Ki 67 siendo débilmente positivo, no se practicó determinación de CD34 en ninguna de las muestras obtenidas.
DiscusiónLa revisión de la clasificación de la OMS agrupa a los gangliogliomas bajo el término de tumores neuronales y glioneurales mixtos en el que se incluyen también el gangliocitoma, el gangliocitoma displásico del cerebelo (enfermedad de Lhermitte-Duclos), astrocitoma/gangliocitoma desmoplásico infantil, tumor disembrioplásico neuroepitelial, neurocitoma central, liponeurocitoma cerebeloso, y paraganglioma del filum3 y a los cuales recientemente se le han añadido una serie de tumores muy poco frecuentes como el tumor papilar glioneural, el tumor glioneural formador de rosetas del IV ventrículo4.
El término ganglioglioma fue introducido por Courville en 19305 para describir un tumor mixto, de muy escasa incidencia, en el que destacaban un componente astrocitario y otro de tipo neural. En la población general representan entre el 0,4-0,7 en el conjunto de todos los tumores cerebrales y hasta el 1% de los tumores medulares; sin embargo, en la edad pediátrica llegan a suponer el 5% de todos los tumores1.
En la mayoría de las series se observa un ligero predominio de los varones sobre las mujeres2,6 y en nuestra casuística existe también un predominio de los varones en relación 1:1,4, sin embargo, en la subserie pediátrica todos los pacientes eran varones y en los adultos existe el doble de mujeres que de varones. Pueden presentarse a cualquier edad y existen casos publicados entre los 2 meses y 80 años, siendo en nuestra experiencia la edad media de 26,4 años en el momento del diagnóstico. No se conocen diferencias geográficas o raciales en cuanto a su incidencia.
En nuestra casuística, al igual que en la mayoría de las series, la forma de presentación más común fue con crisis epilépticas refractarias al tratamiento farmacológico y con un tiempo de evolución muy prolongado2, siendo los déficits neurológicos focales poco frecuentes. En los gangliogliomas de localización en fosa posterior, pueden observarse, al igual que en otros tumores de esta localización, afectación de pares craneales, signos de disfunción cerebelosa, cefalea, etc7. Se han descrito casos de gangliogliomas asociados a otras patologías, como la neurofibromatosis tipo I y II, síndrome de Down, de Turcot, síndrome de Peutz-Jeghers agenesia parcial del cuerpo calloso, polimicrogiria cerebelosa, etc6,8.
Aunque en la descripción inicial de Courville los GG se situaban como tumores predominantes del III ventrículo, topográficamente, pueden localizarse en cualquier punto del SNC, la gran mayoría se sitúan en el lóbulo temporal (17 casos en nuestra serie), pero no son raros en los lóbulos parietal y frontal1,9. En nuestra experiencia no hemos tenido gangliogliomas fuera de los hemisferios cerebrales, se han descrito con menor frecuencia en el cerebelo10, tronco cerebral11, hipófisis12, pineal13, tálamo14 e hipotálamo15, ventrículos supratentoriales y cuarto16, nervios óptico y trigémino17, región suprasellar18, globo ocular19 o en la médula espinal20. La localización infratentorial (tronco, cerebelo y ángulo ponto-cerebeloso) es rara con escasos casos descritos en la literatura21,22. Excepcionalmente pueden ser bilaterales o multicéntricos23.
Microscópicamente se caracterizan por la presencia de neuronas y células gliales maduras generalmente astrocitos pilocíticos o fibrilares y menos frecuentemente oligodendrocítos. La población neuronal está desordenada con variedades morfológicas en su tamaño y con frecuencia se pueden ver células piramidales ganglionares y/o células ganglionares bien diferenciadas. Las mitosis son poco frecuentes y se observan exclusivamente en el componente glial24. Pueden acompañarse de displasia cortical con desorganización cortical, displasias microscópicas glioneurales por alteraciones de la migración neuronal o presentar asociado un componente de xantoastrocitoma pleomórfico25. Excepcionalmente se observan datos de malignidad como anaplasia, proliferación vascular y necrosis. Aunque en nuestra serie únicamente en 9 casos se indica el grado de la OMS encontrándose 6 grados II y 3 grados I, en la literatura existe hasta un 93% grado I, el 6% son grado II y únicamente el 1% son grados III y IV22.
Se han descrito diversas variedades de gangliogliomas como melanóticos26, o microquístico27, así como la asociación con otras neoplasias cerebrales como hemangioma28, xantoastrocitoma pleomorfico29 o con la presencia de otros componentes neoplásicos como oligodendroglial30 como ocurría en nuestro paciente número 12 o presentar componente ependimoma tanacítico31.
Las técnicas inmunohistoquímicas son fundamentales para identificar los dos componentes celulares, así la porción glial muestra positividad para la proteína S-100, proteína gliofibrilar ácida y vimentina, mientras que la población neuronal muestra positividad para la sinaptofisina, enolasa neuronal específica, cromatogranina A, calbindina y calcineurina1,2, hasta el 80% de los GG muestran positividad para el CD342. Algunos biomarcadores como la leptina y su receptor, que se encuentran sobre-expresadas en tumores gliales malignos, tiene una baja expresión en otros de baja agresividad como el ganglioglioma32. En los últimos años se han editado algunos trabajos sobre genética molecular de los GG con la identificación de diversas mutaciones que afectan a los cromosomas, 9, 101,8, y recientemente se ha publicado por primera vez, la presencia de una mutación en el gen NBN en un GG supratentorial extirpado a una niña de 13 años33.
En la tomografía computarizada el 40% son hipodensos, el 30% muestran un quiste hipodenso con un nódulo mural isodenso, el 15% son isodensos y el otro 15% son hiperdensos. Presentan calcificaciones entre un 20 y un 83%. En el 50% de los casos se realzan con contraste34.
Con resonancia magnética se presentan como masas bien delimitadas, manifestándose en T1 como isointensas (40-70%), hipointensas (20-40%) y en T2 hiperintensas (70-90%) o isointensas (20-30%). Se realzan de forma variable con la administración de gadolinio y habitualmente de forma heterogénea24,34. Suelen ejercer poco efecto de masa, se acompañan de escaso edema y excepcionalmente pueden observarse datos de sangrado antiguo. La presencia de edema vasogénico es característica de gangliogliomas de alto grado y en ocasiones pueden observarse áreas de displasia cortical.
En general no es preciso realizar estudio angiográfico, ya que al tratarse de tumores poco vascularizados solo se observan desplazamientos vasculares.
El diagnóstico diferencial radiológico debe realizarse con otras lesiones quísticas intracraneales como los quistes dermoides y epidermoides35, hemagioblastomas quisticos36, y con lesiones tumorales como astrocitoma pilocítico o el ganglioglioma desmoplásico infantil24. En este último, es frecuente observar una lesión quística, generalmente de gran volumen, que puede contener tabiques en su interior y habitualmente en contacto con la duramadre, pudiendo observarse un realce dural24. En espectroscopia por RM muestran un aumento del pico de colina y del índice (Colina/N-acetil aspartato) Cho/NAA, así como una disminución de los índices (Colina/Creatina) Cho/Cr y NAA/Cr, en relación con el mismo área normal del hemisferio contralateral o con los gliomas37.
Recientemente Kikuchi et al38 estudian con RM el coeficiente de difusión aparente en un grupo de 10 pacientes consecutivos con ganglioglioma supratentorial, y lo comparan con otro grupo de tumores astrocitarios de alto y bajo grado, observando que en los GG la media de coeficiente es de 1,45±0,20 x 10−3 mm2/s que es significativamente mayor que el obtenido en astrocitomas de diferentes grados de malignidad. Además, al comparar el CDA con la celularidad de los gangliogliomas se observan que entre ambos existe una relación inversa. En los estudios de PET y SPECT los gangliogliomas de bajo grado muestran una actividad metabólica normal o reducida, mientras que en los de alto grado se observa hiperactividad39.
Se trata de lesiones cuyo tratamiento de primera opción, es la resección quirúrgica completa siempre que esta sea posible, en cuyo caso no sería necesario otros tratamientos complementarios40. El ganglioglioma de bajo grado es poco radiosensible por lo que la radioterapia solo debería emplearse en casos de recidivas no subsidiarias de nueva resección quirúrgica41,42, en aquellos casos en los que se observan astrocitos con de signos de agresividad, o cuando se produce una degeneración maligna. Sin embargo, dado que se trata de una estirpe tumoral de baja malignidad y considerando los efectos deletéreos que la radiación produce sobre la maduración del cerebro infantil, es difícil su justificación actual. Otros aspectos negativos derivados de la radiación serían la inducción de nuevos tumores cerebrales42 o la posibilidad de degeneración maligna. Sin embargo, en la literatura existen casos que han sufrido degeneración maligna sin haber recibido radioterapia2,43.
Recientemente Rades et al44 han realizado una revisión sobre los 402 casos de GG publicados en la literatura, tanto en casos aislados como series amplias. Encontraron que el 15% (60 casos) fueron etiquetados como malignos en función de tener un MIB-1 alto (> 10%) o la presencia de cambios anaplásicos en los tumores. En los 342 casos etiquetados de bajo grado, se realizó exclusivamente tratamiento quirúrgico en 268 pacientes (78,4%) y cirugía complementada con radioterapia en 74 tumores (14 resecciones completas y 60 parciales), es decir el 21,6% de los GG de bajo grado recibieron radioterapia; concluyen la revisión indicando que la resección completa no precisa complementarse con radioterapia y que en los casos de extirpación incompleta, la radioterapia postoperatoria mejora ostensiblemente el control local del tumor. Sin embargo, en esta revisión de Rades et al44 se incluyen gangliogliomas desmoplásicos tanto infantiles como de adultos que son una entidad clínica e histopatológica diferente, por lo que las afirmaciones vertidas deben de ser tomadas con cautela45.
No hay datos concluyentes sobre el beneficio de la quimioterapia46.
El pronóstico es muy favorable cuando se realiza una resección completa con índices de recidiva a los 2 años del 3%. Los pacientes que presentan crisis epilépticas quedan libres de éstas en más del 75% de los casos, pudiéndose controlar mejor en el resto con medicación específica2,47,48. En la revisión de Phi et al49 en la que se analizan 87 pacientes con glioma de bajo grado que debutaron con epilepsia, el control postquirúrgico de las crisis mejora con el paso del tiempo, obteniendo en el primer año un control del 79%, pasando al 86% en el segundo y del 92% en el quinto año. En los tumores de localización infratentorial debe tenerse especial cuidado con la posibilidad de provocar déficits neurológicos y en el caso de quedar restos tumorales su comportamiento es más benigno que el de otros tumores de la misma localización21. En los casos de recidiva es frecuente observar un incremento en el grado de malignidad a expensas del componente glial.
La reducción o supresión de los fármacos antiepilépticos en general está poco estudiado en la literatura, en nuestra experiencia 5 pacientes (27,7%) no precisaban medicación a los 2 años de la cirugía. En la serie de Zaatreh et al50 sobre 67 pacientes con epilepsia tumoral del lóbulo temporal, únicamente 17 pacientes (28% del total), de los 44 que quedaron libres de crisis llegaron a suspender totalmente la medicación. En la serie pediátrica de Iannelli et al51 el porcentaje de niños libres de medicación es únicamente del 16%. Sorpresivamente en la reciente publicación de Phi et al49 el porcentaje alcanza el 89% que los autores atribuyen a los diferentes métodos estadísticos empleados, a los diversos sistemas de control utilizados o las diferentes características de las series analizadas ya que el grado de suspensión de la medicación es más alto en los pacientes pediátricos que en los adultos, aunque esta afirmación no está avalada en otras referencias bibliográficas51; otro factor a tener en cuenta cuando se analiza este hecho es en nuestra opinión, la heterogeneidad de las series publicadas con diferentes edades, localizaciones, tipos tumorales, etc.
Se consideran factores de buen pronóstico y por lo tanto con menor tasa de recidiva los grados I, los localizados en el lóbulo temporal, los que debutan con epilepsia de larga evolución y cuando se realiza una extirpación completa evidenciada en resonancia magnética2,52–54. La edad del paciente en el momento de la cirugía no tiene valor pronostico2.
Como factores de mal pronóstico, además del mayor grado histológico se debe incluir la presencia de atipia celular10,53, la localización en la línea media10 y la resección incompleta aunque en la series de Lang et al55 y de Rumana et al56 no mencionan este hecho, pero hay que tener en cuenta que estos autores no mencionan los estudios de RM postoperatoria.
En nuestra serie no se realizó en ningún caso determinación del Ki-67 y de la p53 aunque estos parámetros pueden tener un importante valor pronóstico como demostraron Hirose et al57 al observar cifras significativamente más altas en los casos de recidiva en relación a los no recurrentes. La diseminación a través del líquido cefalorraquídeo es poco frecuente. En la serie de Hukim et al58 sobre 96 gangliogliomas observan 4 casos y de estos únicamente 2 eran de hemisferios cerebrales, siendo los otros de tronco cerebral y médula espinal.
Los gangliogliomas anaplásicos se comportan de una forma más agresiva tanto a nivel local como ocasionando siembras leptomeníngeas, aunque la existencia de esta diseminación para Hukin et al58 no supone en principio un peor pronóstico comparado con otros tumores de malignidad superponible. En los gangliogliomas anaplásicos o recidivados la evidencia con técnicas inmunohistoquímicas de la proteína “antiapoptótica survivin”, que muestran más de un 5% de las células gliales, se considera como un factor de mal pronóstico59.
Podemos concluir, aunque nuestro estudio se encuentra limitado por el número de casos que no permite la realización de un estudio estadístico riguroso.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

