



El insomnio fatal esporádico (IFE) es una patología muy infrecuente que comparte características clínicas con la forma familiar. Su sintomatología es muy heterogénea, destacando una rápida progresión y una corta esperanza de vida. El diagnóstico definitivo es anatomopatológico, debiendo descartarse la mutación D178N del gen de la proteína priónica1,2. Presentamos el caso de un paciente que debutó con un fenotipo sugerente de parálisis supranuclear progresiva (PSP) de rápida evolución.
Caso clínicoVarón de 50 años; su padre había fallecido por una probable demencia con cuerpos de Lewy en una edad senil y no tenía otros antecedentes familiares de interés. Entre sus antecedentes personales destacaba únicamente una sífilis con manifestación cutánea tratada con penicilina.
La sintomatología inicial fue de índole neuropsiquiátrica: depresión, aislamiento social y sentimiento de incapacidad, con menoscabo en su vida personal y laboral. La primera atención por parte de Neurología fue motivada por la aparición insidiosa de diplopía binocular. En los siguientes meses la progresión fue rápida: lentitud motora con afectación de la destreza manipulativa, sensación de inestabilidad con caídas frecuentes y alteración del habla con cambio en el tono de la voz. Los familiares apreciaron significativos cambios en su personalidad y el propio paciente reconocía fallos de memoria y de concentración. A la anamnesis para síntomas disautonómicos se recogió únicamente disfunción eréctil. No presentaba trastornos del sueño.
Se describe a continuación la exploración física realizada unos diez meses después del debut clínico. No se encontraron signos patológicos en la exploración general. A nivel neuropsicológico destacaba una afectación de las funciones ejecutivas y de la memoria, sin apraxias ni alteración visuoespacial, con reflejos de liberación frontal presentes (Montreal cognitive assessment: 23/30). El lenguaje era fluente, sin rasgos disfásicos, y el habla mostraba una disprosodia con componente escandido. Existía limitación de la mirada vertical, alteración del seguimiento ocular y sacadas en forma de «ondas cuadradas». A nivel motor presentaba bradicinesia generalizada con rigidez de predominio axial, sin temblor ni mioclonías, tampoco fasciculaciones ni afectación del aspecto trófico. El balance muscular era normal. Los reflejos de estiramiento muscular estaban exaltados en todos los grupos, con signo de Hoffman bilateral y respuesta flexora cutaneoplantar. La exploración de la sensibilidad superficial y profunda era normal. Las pruebas de coordinación y de la marcha demostraban dismetría de las cuatro extremidades, ampliación de la base en la bipedestación y desequilibrio durante la deambulación.
Nos encontramos ante un trastorno neurológico multisistémico rápidamente progresivo. Las etiologías neurodegenerativa, disinmune y priónica constituyeron el principal diagnóstico diferencial. La PSP fue la primera sospecha al comienzo del cuadro. En cuanto a entidades inmunomediadas, las encefalitis por anticuerpos contra IgLON5, DPPX, mGluR1 y GAD65 fueron las consideradas más probables. Otras enfermedades que también se tuvieron en cuenta fueron la neurosífilis, la enfermedad de Niemann-Pick tipoC, la enfermedad de Whipple y las ataxias heredodegenerativas.
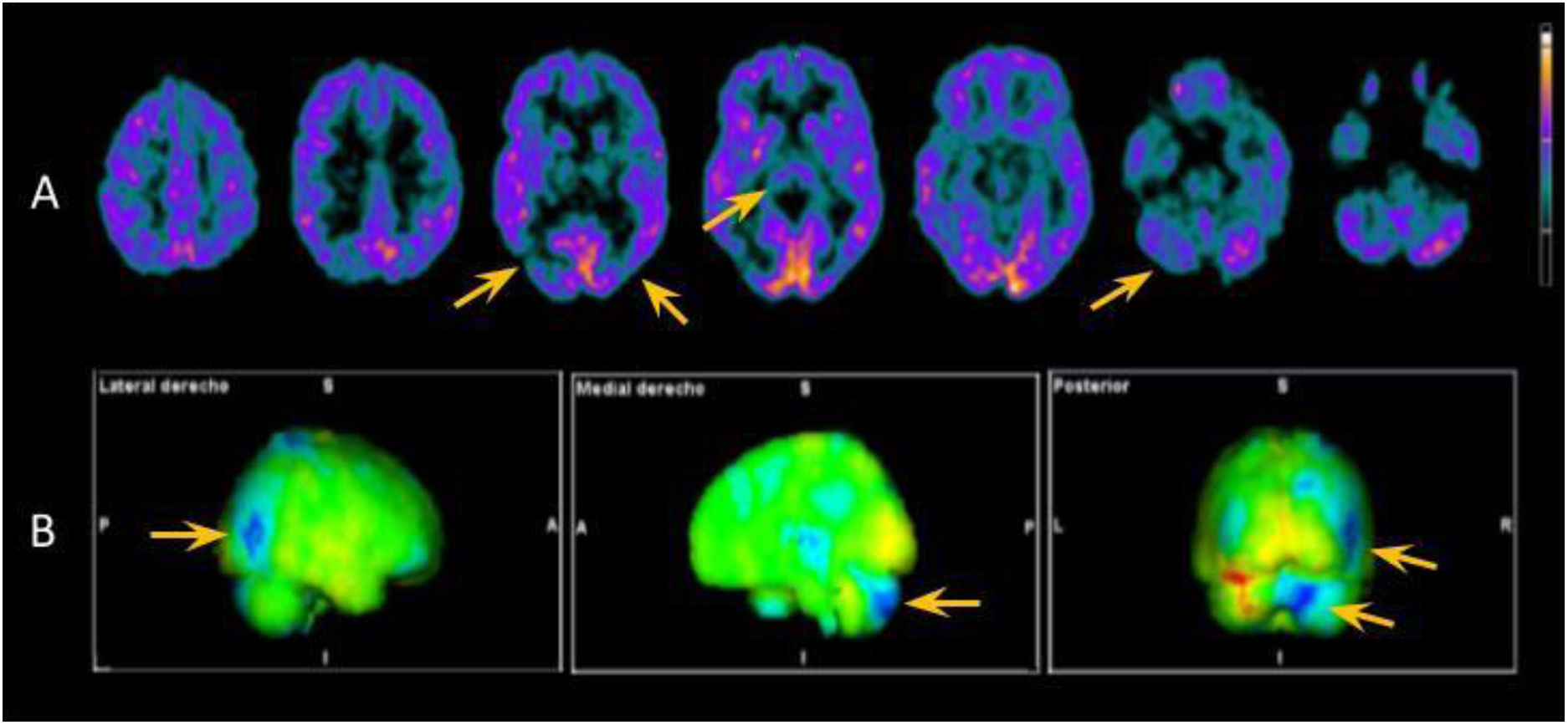
Atendiendo a estas posibilidades, se llevó a cabo un amplio estudio complementario. La resonancia magnética (RM) cerebral no mostró hallazgos significativos. En el líquido cefalorraquídeo (LCR) existía una hiperproteinorraquia (66mg/dl [normal: <45mg/dl]) sin pleocitosis ni consumo de glucosa, con un estudio microbiológico negativo (incluyendo VDRL y PCR de Tropheryma whipplei) y un análisis citológico normal. La autoinmunidad en el LCR y en el suero fue negativa, incluyendo los anticuerpos contra IgLON5, CASPR2, DPPX, mGluR1, GAD65, AMPAr, NMDAr, GABAr y LGI1. Se detectó una elevación de la proteína tau total, con un valor de 467pg/ml (patológico para su edad >116pg/ml), con valores normales de beta-amiloide y tau fosforilada. No se detectaron proteína 14-3-3 ni proteína priónica mediante RT-QuIC en el LCR. Se realizaron varios electroencefalogramas en vigilia que no mostraron ningún patrón patológico. Sí fueron patológicas la PET-[18F]fludeoxiglucosa cerebral (fig. 1) y la SPECT-[123I]ioflupano (hipocaptación simétrica de ambos núcleos estriados).
![Exploración de medicina nuclear mediante PET [18F]FDG. A)Cortes axiales seleccionados en los que se visualiza hipometabolismo en tálamo, encrucijada temporo-parieto-occipital bilateral de predominio derecho y en el hemisferio cerebeloso derecho (flechas amarillas). B)Imágenes proyectadas mediante mapas 3D-stereotactic surface projection que comparan los hallazgos anteriores con una base de pacientes normales de acuerdo con la edad, en las que se confirma lo anteriormente descrito. Las regiones hipometabólicas se representan en color azul.](https://static.elsevier.es/multimedia/02134853/unassign/S0213485324000586/v1_202402060423/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9i85vcbshWVQPfl+0x3Npqwg49cnR8y8IIj2HMmKNuXXh1W7/MTWUcrRXrO5++nRmIgSf5m92znmx4vPEcuGzsb/sREa2Rb7X9ZpoICIJVUNNR2rHsjdIhfbWadWYWOQN1sWYSgnjfllZ82ZF9uLpNRrOfaaAPlgQC5GSNvGinv0h5jvDu3CM1mbm2rFXSY+Yv0G1FneEGZumlqw9zs1EPeik78RTHj/FgDkybvDXru4c= "Exploración de medicina nuclear mediante PET [18F]FDG. A)Cortes axiales seleccionados en los que se visualiza hipometabolismo en tálamo, encrucijada temporo-parieto-occipital bilateral de predominio derecho y en el hemisferio cerebeloso derecho (flechas amarillas). B)Imágenes proyectadas mediante mapas 3D-stereotactic surface projection que comparan los hallazgos anteriores con una base de pacientes normales de acuerdo con la edad, en las que se confirma lo anteriormente descrito. Las regiones hipometabólicas se representan en color azul.")
Exploración de medicina nuclear mediante PET [18F]FDG. A)Cortes axiales seleccionados en los que se visualiza hipometabolismo en tálamo, encrucijada temporo-parieto-occipital bilateral de predominio derecho y en el hemisferio cerebeloso derecho (flechas amarillas). B)Imágenes proyectadas mediante mapas 3D-stereotactic surface projection que comparan los hallazgos anteriores con una base de pacientes normales de acuerdo con la edad, en las que se confirma lo anteriormente descrito. Las regiones hipometabólicas se representan en color azul.
Durante el seguimiento se realizaron ensayos terapéuticos con corticoides a dosis altas, inmunoglobulinas intravenosas y levodopa (prueba aguda con 250mg y mantenimiento con 100mg cada 8horas), con resultados infructuosos. El estado clínico continuó empeorando: mayor afectación cognitiva, ataxia grave con incapacidad para la deambulación autónoma y disfagia severa. Aproximadamente un año y medio después del inicio de la sintomatología falleció como consecuencia de una infección respiratoria.
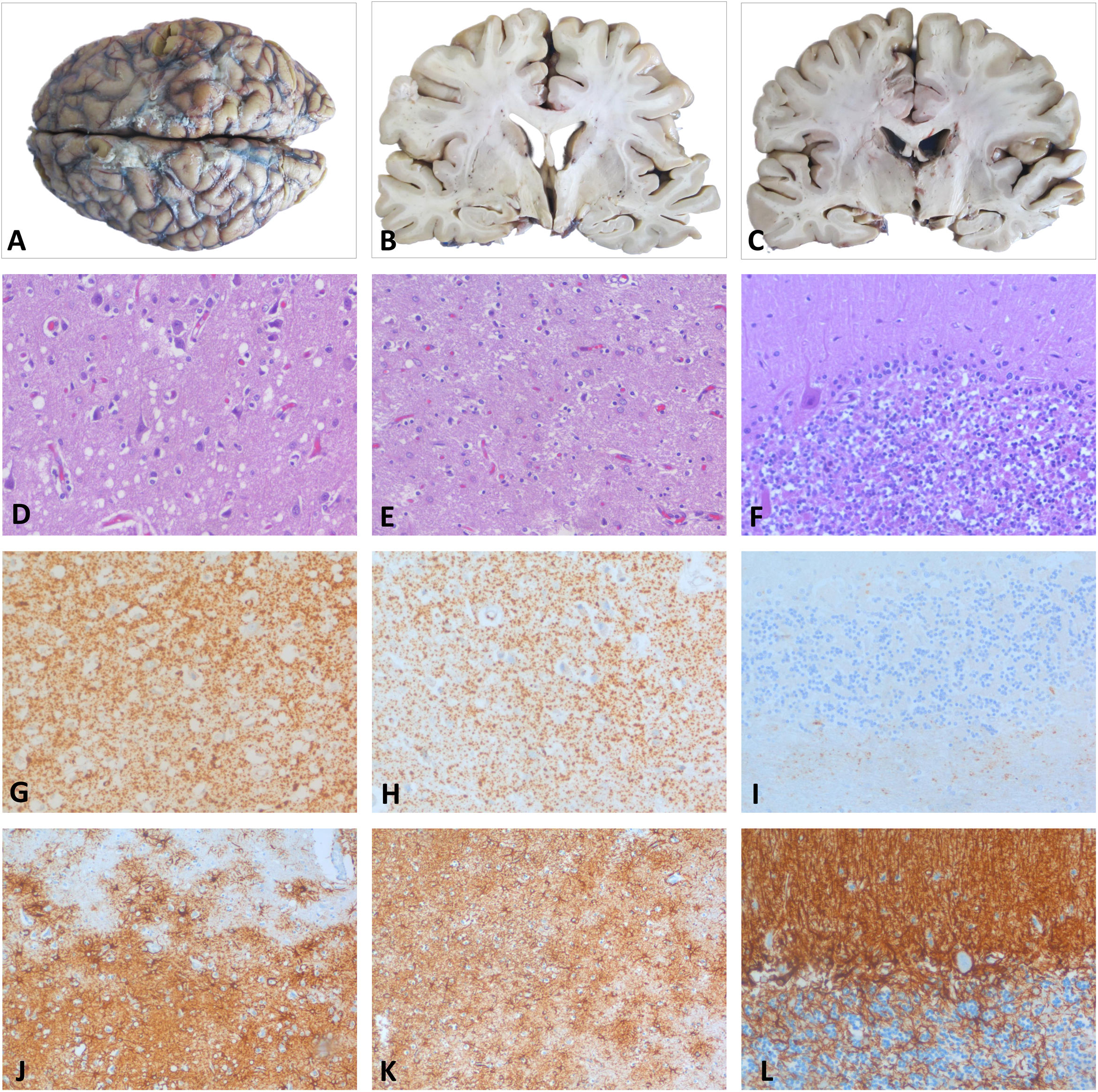
DiagnósticoEl estudio neuropatológico post mortem reveló cambios neurodegenerativos propios del insomnio fatal, caracterizados por la clásica atrofia y gliosis astrocitaria reactiva de tálamo y núcleos olivares inferiores, junto a cambios espongiformes de vacuola pequeña en la corteza cerebral y depósito de proteína priónica de tipo difuso-sináptico. En el cerebelo, la afectación era predominante en el vermis y en la corteza hemisférica derecha, existiendo una moderada pérdida celular con focos difusos de proteína priónica y gliosis (fig. 2)3,4. El análisis genético y molecular descartó la mutación D178N y demostró una forma MM2 del codón129, llegando al diagnóstico definitivo de IFE5-10.
Estudio macroscópico: encéfalo y cortes coronales a nivel de ganglios basales y tálamo que revela una mínima atrofia en la corteza temporal sin otros cambios relevantes. Córtex parietal (D, G y J): pérdida neuronal y moderada espongiosis del neuropilo con vacuolas redondeadas de pequeño-mediano tamaño con HE (D). El estudio inmunohistoquímico (IHQ) reveló un depósito de la PrP con patrón granular-sináptico difuso (G) y una marcada gliosis astrocitaria reactiva con la GFAP (J). Tálamo (E, H y K): pérdida neuronal con mínima espongiosis y severa gliosis astrocitaria con la HE (E) y GFAP (K). Presencia de proteína priónica de forma más discreta que en la corteza (H). Cerebelo (F, I y L): moderada pérdida de neuronas granulares y de células de Purkinje con la HE (F) con presencia de pequeños focos de PrP en la capa de células granulares (I) y gliosis astrocitaria con la GFAP (L). GFAP: proteína ácida fibrilar glial; HE: hematoxilina-eosina; PrP: proteína priónica.")
Histopatología post mortem. A-C)Estudio macroscópico: encéfalo y cortes coronales a nivel de ganglios basales y tálamo que revela una mínima atrofia en la corteza temporal sin otros cambios relevantes. Córtex parietal (D, G y J): pérdida neuronal y moderada espongiosis del neuropilo con vacuolas redondeadas de pequeño-mediano tamaño con HE (D). El estudio inmunohistoquímico (IHQ) reveló un depósito de la PrP con patrón granular-sináptico difuso (G) y una marcada gliosis astrocitaria reactiva con la GFAP (J). Tálamo (E, H y K): pérdida neuronal con mínima espongiosis y severa gliosis astrocitaria con la HE (E) y GFAP (K). Presencia de proteína priónica de forma más discreta que en la corteza (H). Cerebelo (F, I y L): moderada pérdida de neuronas granulares y de células de Purkinje con la HE (F) con presencia de pequeños focos de PrP en la capa de células granulares (I) y gliosis astrocitaria con la GFAP (L).
GFAP: proteína ácida fibrilar glial; HE: hematoxilina-eosina; PrP: proteína priónica.
Exponemos en este trabajo un caso diagnosticado de IFE a partir de unos resultados anatomopatológico, genético y molecular concluyentes. La sintomatología predominante fue cognitiva y motora en forma de PSP-like con ataxia grave y evolución atípica, sin alteración manifiesta del sueño, lo que, sumado a los resultados negativos de la proteína 14-3-3 y del RT-QuIC, dificultaron pre mortem pensar en el diagnóstico de una encefalopatía priónica tipo insomnio fatal11-13. En varias revisiones previas se han obtenido resultados similares de los análisis de la 14-3-3, el RT-QuIC y la proteína tau en el LCR, lo que diferencia el insomnio fatal de otras prionopatías más frecuentes2,14. Por otro lado, las exploraciones de medicina nuclear del cerebro fueron relevantes en comparación a los hallazgos poco significativos de la RM encefálica. El hipometabolismo talámico es un dato destacable que puede ayudar a considerar esta enfermedad en los cuadros clínicos compatibles12,15. En nuestro caso no se realizó una polisomnografía por no haber presentado clínica específica, pero podría haber aportado datos de utilidad en el proceso diagnóstico2,8.
El insomnio fatal, tanto en su forma esporádica como en la familiar, supone un reto clínico por su infrecuencia y por los hallazgos poco definitorios de las pruebas complementarias. Debe considerarse esta posibilidad en aquellos sujetos que sufran un trastorno neurológico multisistémico de rápida evolución con resultados negativos de las pruebas complementarias. En estos, la PET-[18F]fludeoxiglucosa cerebral puede ser útil para orientar el diagnóstico. Ponemos en valor la importancia de realizar estudios anatomopatológicos post mortem en aquellos casos neurológicos complejos e inconclusos.
Consideraciones éticasEste trabajo cumple los requerimientos éticos exigidos por nuestro centro hospitalario.
Consentimiento informadoConsentimiento obtenido para la publicación del caso.
FinanciaciónNinguna.
Conflicto de interesesNinguno.

