




El objetivo de este estudio fue analizar el proceso diagnóstico de los pacientes con fibrosis pulmonar idiopática en España, desde el inicio de los síntomas hasta el diagnóstico y tratamiento antifibrótico, en relación con la publicación de las sucesivas guías de práctica clínica.
Material y métodosEstudio multicéntrico, observacional, ambispectivo, en el que se analizaron los pacientes incluidos en el registro de la fibrosis pulmonar idiopática de la Sociedad Española de Neumología y Cirugía Torácica. Para ello se habilitó un cuaderno electrónico de recogida de datos en la web de la sociedad. Se recogieron variables sociodemográficas y clínicas al diagnóstico y seguimiento de los pacientes.
ResultadosDesde enero de 2012 hasta diciembre de 2019 se incluyeron 1.064 pacientes, siendo finalmente analizados 929. El proceso diagnóstico varió en función del año en el que se realizó el diagnóstico y el patrón radiológico observado en la tomografía computarizada de alta resolución. En 244 (26,3%) pacientes, el diagnóstico se realizó con tomografía computarizada de alta resolución de tórax y evaluación clínica. La biopsia quirúrgica se utilizó hasta en el 50,2% de los casos diagnosticados antes del 2011, y en un 14,2% a partir de 2018. La mediana de tiempo que transcurre desde el inicio de los síntomas hasta el diagnóstico es de 360 días (RIC 120-720), siendo mayor de 2 años en el 21,0% de los pacientes. Recibieron tratamiento antifibrótico al 79,4% de los pacientes. El tiempo desde el diagnóstico hasta el inicio del tratamiento fue de 309±596,5 días, con una mediana de 49 (RIC 0-307).
ConclusionesEl proceso diagnóstico, incluyendo el tiempo hasta el diagnóstico y el tipo de pruebas utilizadas, ha ido cambiando desde 2011 hasta 2019, probablemente debido el avance en la investigación clínica y la publicación de guías consenso diagnóstico-terapéuticas.
The objective of the study was to analyze the diagnostic process and the time until the start of treatment of patients with idiopathic pulmonary fibrosis in relation to the publication of successive clinical practice guide.
Material and methodsMulticenter, observational, ambispective study, in which patients includes in the idiopathic pulmonary fibrosis registry of the Spanish Society of Pulmonologist and Thoracic Surgery were analyzed. An electronic data collection notebook was enabled on the society's website. Sociodemographic and clinical variables were collected at diagnosis and follow-up of the patients.
ResultsFrom January 2012 to december 2019, 1064 patients were included in the registry, with 929 finally analyzed. The diagnosis process varied depending on the year in which it was performed, and the radiological pattern observed in the high-resolution computed tomography. Up to 26.3% of the cases (244) were diagnosed with chest high-resolution computed tomography and clinical evaluation. Surgical biopsy was used up to 50.2% of cases diagnosed before 2011, while it has been used in 14.2% since 2018. The median time from the onset of symptoms to diagnosis was 360 days (IQR 120-720), taking more than 2 years in the 21.0% of patients. A percentage of 79.4 of patients received antifibrotic treatment. The average time from diagnosis to the antifibrotic treatment has been 309±596.5 days, with a median of 49 (IQR 0-307).
ConclusionsThe diagnostic process, including the time until diagnosis and the type of test used, has changed from 2011 to 2019, probably due to advances in clinical research and the publication of diagnostic-therapeutic consensus guidelines.
La fibrosis pulmonar idiopática (FPI) es la enfermedad pulmonar intersticial difusa (EPID) más frecuente1–3; se trata de una enfermedad grave, progresiva y mortal con una supervivencia media estimada de 3 a 5 años sin tratamiento antifibrótico. Definida como nueva entidad en 1970 por Scadding4, ha estado en continuo proceso de caracterización diagnóstica durante los últimos 20 años.
Las guías internacionales ATS/ERS/JRS5–8 han permitido una mejor aproximación diagnóstica y terapéutica, definiendo los hallazgos de la tomografía axial computarizada de alta resolución (TACAR) y su correlación con la anatomía patológica, enfatizando el papel del comité multidisciplinar (CMD) como el estándar de oro para su diagnóstico. Este enfoque multidisciplinario ha demostrado tener muchos beneficios para la atención clínica de estos pacientes, mejorando la confianza del diagnóstico y guiando las decisiones de tratamiento9. Fue la guía de 20115 la primera en definir el «patrón NIU» en la TACAR y permitir el diagnóstico clínico-radiológico de FPI entre los pacientes con este patrón radiológico, evitando así la necesidad de realizar una biopsia quirúrgica a todos los pacientes. En el 2015 se realizó una actualización del tratamiento7, y en el 20188 de nuevo se reevaluaron los patrones radiológicos, histológicos y el papel de las técnicas diagnósticas.
El tiempo que transcurre desde el inicio de los síntomas hasta que el paciente es diagnosticado es una variable asociada a una mayor mortalidad y a un intervalo libre de progresión más breve10–13. Este periodo de tiempo puede ser superior a 3 años en uno de cada 4 pacientes11. Actualmente, con la existencia de fármacos antifibróticos que enlentecen la progresión de la enfermedad14,15, el diagnóstico precoz de la FPI cobra especial interés.
El objetivo de este estudio fue analizar el proceso diagnóstico de los pacientes con FPI desde el inicio de los síntomas hasta el diagnóstico definitivo, así como el tiempo que transcurre desde el diagnóstico hasta el inicio del tratamiento antifibrótico, en relación con la publicación de las sucesivas guías internacionales de práctica clínica.
Material y métodosDiseñoEstudio ambispectivo, observacional, multicéntrico en el que se analizaron los pacientes incluidos en el registro FPI de la Sociedad Española de Neumología y Cirugía Torácica (SEPAR), entre enero de 2012 y diciembre de 2019. Se invitó a participar en el registro a todos los miembros del área de EPID, mediante un correo electrónico. Para la inclusión de casos, se habilitó un cuaderno de recogida de datos en la web de la SEPAR.
La conducción del estudio siguió en todo momento la normativa de buenas prácticas clínicas y la Declaración de Helsinki. Cada hospital participante obtuvo la aprobación del comité de ética.
PoblaciónLa población a estudio16 fueron sujetos con el diagnóstico de FPI en seguimiento en consultas externas de los centros participantes: se incluyeron tanto los pacientes existentes como los nuevos diagnósticos. Los criterios de inclusión fueron los siguientes: diagnóstico de FPI según las guías clínicas vigentes en ese momento5,6; tener más de 18 años; firmar el consentimiento informado. Los criterios de exclusión fueron: ser menor de 18 años; no firmar el consentimiento informado o ser incapaz de darlo por minusvalía psíquica o física; pacientes diagnosticados de otro EPID fibrosante no FPI.
Procedimientos y variablesSe recogieron variables sociodemográficas y clínicas al diagnóstico, incluyendo antecedentes, síntomas, analítica, patrón de la TACAR de tórax, pruebas de función respiratoria, resultados obtenidos en los procedimientos diagnósticos complementarios como el lavado broncoalveolar (LBA), la biopsia transbronquial por criobiopsia (en adelante criobiopsia) o la biopsia pulmonar por videotoracoscopia, discusión en el CMD y tratamiento administrado. Como otra variable se incluyó el nivel de acreditación SEPAR de las unidades de EPID de los centros participantes (unidades de alta complejidad, unidades especializadas y unidades básicas).
Los pacientes fueron divididos en 4 grupos según su fecha de diagnóstico, de acuerdo con la fecha de publicación de las 3 guías internacionales publicadas durante el periodo del estudio: Grupo A, pacientes diagnosticados antes de la publicación de la guía internacional de 20115; Grupo B, pacientes diagnosticados entre la guía de 20115 y la guía de 20157; Grupo C, pacientes diagnosticados entre la guía de 20157 y la de 20187; Grupo D, pacientes diagnosticados tras la guía de 20188.
Para conocer el tiempo hasta el diagnóstico se tuvo en cuenta la duración de los síntomas del paciente y las fechas de las pruebas complementarias utilizadas para el diagnóstico o la fecha de diagnóstico multidisciplinar. También se analizó el periodo transcurrido desde el diagnóstico hasta el inicio del tratamiento antifibrótico. Es importante señalar que en España el tratamiento antifibrótico con pirfenidona fue financiado por el Sistema Nacional de Salud a partir de septiembre de 2014 en pacientes con FPI con porcentaje de FVC del 50-80% sobre el teórico, y el tratamiento con nintedanib a partir de enero de 2015. El tratamiento de pacientes con FVC superior al 80% no fue posible hasta noviembre de 2016. Los pacientes que comenzaron tratamiento con los antifibróticos antes de la fecha de financiación lo hicieron a través de ensayos clínicos o programas especiales de acceso al medicamento.
Análisis estadísticoSe realizó un análisis estadístico descriptivo utilizando frecuencias y porcentajes para las variables categóricas y la media con desviación típica o mediana y el rango intercuartílico (RIC) para las variables continuas.
Para medir la asociación entre las variables categóricas y el tiempo hasta el diagnóstico se usó la prueba de la Chi-cuadrado o el test exacto de Fisher. En cambio, para la comparación de medias se utilizó la t de Student (o Wilcoxon en caso de que no pudiese asumirse normalidad). Todos los cálculos estadísticos se realizaron mediante el programa SAS System v. 9.4. El nivel de significación se asumió cuando p<0,05.
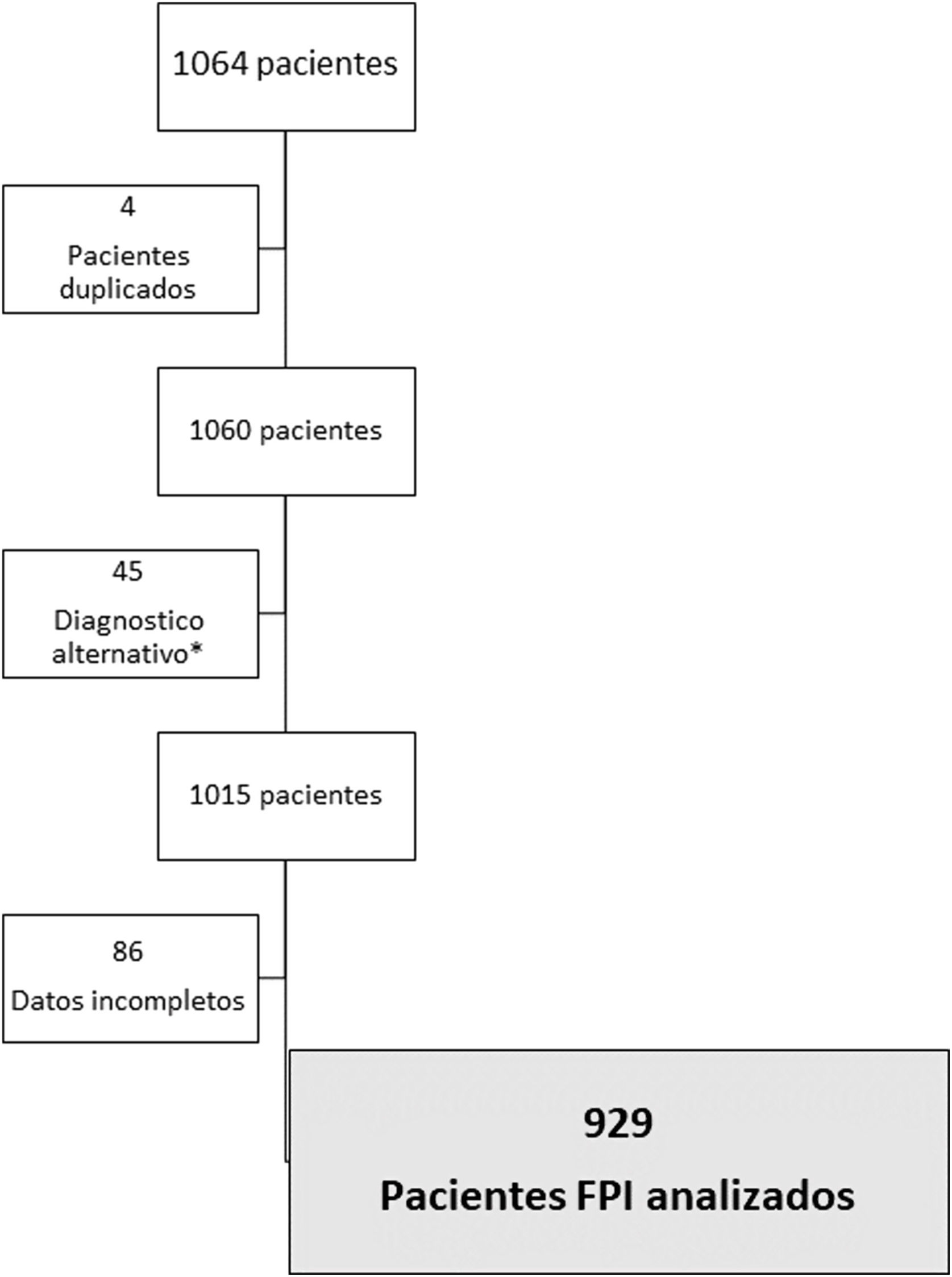
ResultadosDesde enero de 2012 hasta diciembre de 2019 se incluyeron 1.064 pacientes, de los cuales han sido analizados en el presente estudio 929. En la figura 1 se muestra el diagrama de flujo de los pacientes incluidos y las razones de su exclusión. Cuarenta y seis unidades de EPID de diferentes hospitales distribuidos por toda la geografía española participaron en el registro. Veinte de estas estaban acreditadas por SEPAR como unidades de «alta complejidad» (n=14), «especializadas» (n=4) o «básicas» (n=2), incluyendo entre todas ellas el 75,7% del total de los pacientes.

Diagrama de flujo de los pacientes incluidos en el estudio y las causas de su exclusión. *Los diagnósticos alternativos declarados durante el seguimiento de los pacientes fueron: 16 posibles neumonitis por hipersensibilidad crónica, una NINE fibrótica, 4 enfermedades autoinmunes y dudas diagnósticas por falta de progresión de la enfermedad en el paciente 24.
Seiscientos veintisiete (67,5%) de los casos incluidos en el Registro FPI de SEPAR eran varones fumadores o exfumadores y 98 (10,5%) pacientes presentaban antecedentes familiares de EPID. Las comorbilidades declaradas más frecuentemente fueron la hipertensión arterial (n=263; 28,3%), la diabetes mellitus (n=151; 16,3%), la cardiopatía isquémica (n=86; 9,3%), la enfermedad vascular periférica (n=39; 4,2%), la enfermedad cerebrovascular (n=30; 3,2%) y 55 (5,9%) sujetos incluidos habían presentado una neoplasia de órgano sólido en los 5 años previos al diagnóstico. Además, 279 (30%) de los pacientes estaba en tratamiento con estatinas, 151 (16,3%) con ansiolíticos/antidepresivos, 313 (33,7%) con antiagregantes y 63 (6,8%) con anticoagulantes. Sin embargo, en 214 (22,8%) pacientes no se declaró ninguna comorbilidad.
En la tabla 1 se describen las características sociodemográficas y clínicas de los pacientes, subdivididas en grupos según el año de diagnóstico, y en la tabla 2, en función del método diagnóstico utilizado. Desde el punto de vista funcional, en el momento del diagnóstico 50 (5,3%) pacientes presentan una FVC≤50%, 455 (48,6%) una FVC entre 80-51% y 431 (46,0%) una FVC>80%. El diagnóstico se realizó mediante solo una TACAR de tórax e historia clínica compatible en 244 (26,3%) de los pacientes; 210 (22,6%) requirieron además de un LBA; 106 sujetos (11,4%) biopsia quirúrgica y LBA; 53 pacientes (5,7%) LBA y criobiopsia; 7 pacientes (0,7%) sólo criobiopsia; 108 (11,6%) sólo biopsia quirúrgica, y finalmente, 15 (1,6%) precisaron LBA, criobiopsia y biopsia quirúrgica.
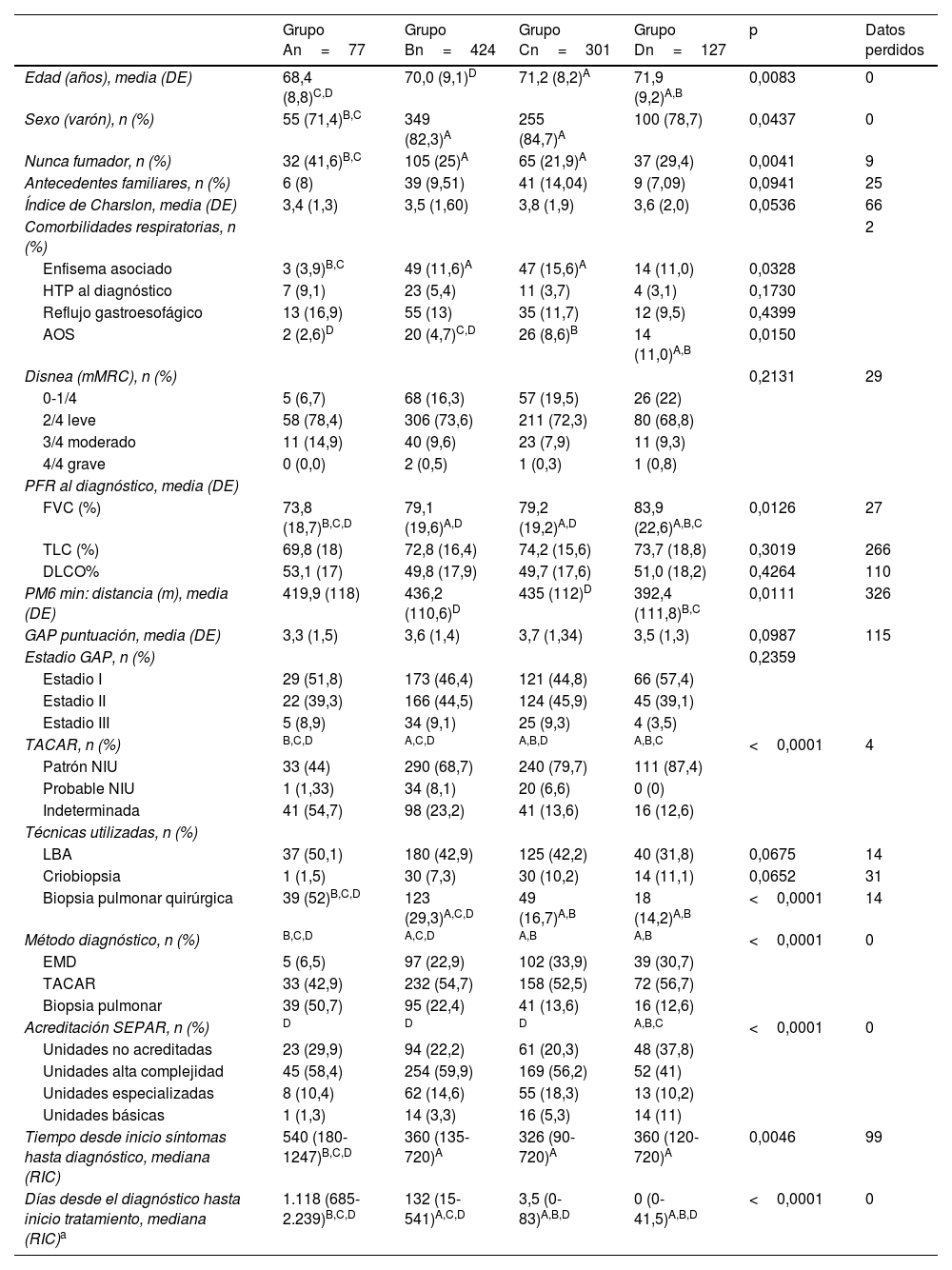
Características sociodemográficas y clínicas de los pacientes con FPI incluidos en el Registro FPI de SEPAR, en función de la fecha de diagnóstico y guía clínica internacional vigente
| Grupo An=77 | Grupo Bn=424 | Grupo Cn=301 | Grupo Dn=127 | p | Datos perdidos | |
|---|---|---|---|---|---|---|
| Edad (años), media (DE) | 68,4 (8,8)C,D | 70,0 (9,1)D | 71,2 (8,2)A | 71,9 (9,2)A,B | 0,0083 | 0 |
| Sexo (varón), n (%) | 55 (71,4)B,C | 349 (82,3)A | 255 (84,7)A | 100 (78,7) | 0,0437 | 0 |
| Nunca fumador, n (%) | 32 (41,6)B,C | 105 (25)A | 65 (21,9)A | 37 (29,4) | 0,0041 | 9 |
| Antecedentes familiares, n (%) | 6 (8) | 39 (9,51) | 41 (14,04) | 9 (7,09) | 0,0941 | 25 |
| Índice de Charslon, media (DE) | 3,4 (1,3) | 3,5 (1,60) | 3,8 (1,9) | 3,6 (2,0) | 0,0536 | 66 |
| Comorbilidades respiratorias, n (%) | 2 | |||||
| Enfisema asociado | 3 (3,9)B,C | 49 (11,6)A | 47 (15,6)A | 14 (11,0) | 0,0328 | |
| HTP al diagnóstico | 7 (9,1) | 23 (5,4) | 11 (3,7) | 4 (3,1) | 0,1730 | |
| Reflujo gastroesofágico | 13 (16,9) | 55 (13) | 35 (11,7) | 12 (9,5) | 0,4399 | |
| AOS | 2 (2,6)D | 20 (4,7)C,D | 26 (8,6)B | 14 (11,0)A,B | 0,0150 | |
| Disnea (mMRC), n (%) | 0,2131 | 29 | ||||
| 0-1/4 | 5 (6,7) | 68 (16,3) | 57 (19,5) | 26 (22) | ||
| 2/4 leve | 58 (78,4) | 306 (73,6) | 211 (72,3) | 80 (68,8) | ||
| 3/4 moderado | 11 (14,9) | 40 (9,6) | 23 (7,9) | 11 (9,3) | ||
| 4/4 grave | 0 (0,0) | 2 (0,5) | 1 (0,3) | 1 (0,8) | ||
| PFR al diagnóstico, media (DE) | ||||||
| FVC (%) | 73,8 (18,7)B,C,D | 79,1 (19,6)A,D | 79,2 (19,2)A,D | 83,9 (22,6)A,B,C | 0,0126 | 27 |
| TLC (%) | 69,8 (18) | 72,8 (16,4) | 74,2 (15,6) | 73,7 (18,8) | 0,3019 | 266 |
| DLCO% | 53,1 (17) | 49,8 (17,9) | 49,7 (17,6) | 51,0 (18,2) | 0,4264 | 110 |
| PM6 min: distancia (m), media (DE) | 419,9 (118) | 436,2 (110,6)D | 435 (112)D | 392,4 (111,8)B,C | 0,0111 | 326 |
| GAP puntuación, media (DE) | 3,3 (1,5) | 3,6 (1,4) | 3,7 (1,34) | 3,5 (1,3) | 0,0987 | 115 |
| Estadio GAP, n (%) | 0,2359 | |||||
| Estadio I | 29 (51,8) | 173 (46,4) | 121 (44,8) | 66 (57,4) | ||
| Estadio II | 22 (39,3) | 166 (44,5) | 124 (45,9) | 45 (39,1) | ||
| Estadio III | 5 (8,9) | 34 (9,1) | 25 (9,3) | 4 (3,5) | ||
| TACAR, n (%) | B,C,D | A,C,D | A,B,D | A,B,C | <0,0001 | 4 |
| Patrón NIU | 33 (44) | 290 (68,7) | 240 (79,7) | 111 (87,4) | ||
| Probable NIU | 1 (1,33) | 34 (8,1) | 20 (6,6) | 0 (0) | ||
| Indeterminada | 41 (54,7) | 98 (23,2) | 41 (13,6) | 16 (12,6) | ||
| Técnicas utilizadas, n (%) | ||||||
| LBA | 37 (50,1) | 180 (42,9) | 125 (42,2) | 40 (31,8) | 0,0675 | 14 |
| Criobiopsia | 1 (1,5) | 30 (7,3) | 30 (10,2) | 14 (11,1) | 0,0652 | 31 |
| Biopsia pulmonar quirúrgica | 39 (52)B,C,D | 123 (29,3)A,C,D | 49 (16,7)A,B | 18 (14,2)A,B | <0,0001 | 14 |
| Método diagnóstico, n (%) | B,C,D | A,C,D | A,B | A,B | <0,0001 | 0 |
| EMD | 5 (6,5) | 97 (22,9) | 102 (33,9) | 39 (30,7) | ||
| TACAR | 33 (42,9) | 232 (54,7) | 158 (52,5) | 72 (56,7) | ||
| Biopsia pulmonar | 39 (50,7) | 95 (22,4) | 41 (13,6) | 16 (12,6) | ||
| Acreditación SEPAR, n (%) | D | D | D | A,B,C | <0,0001 | 0 |
| Unidades no acreditadas | 23 (29,9) | 94 (22,2) | 61 (20,3) | 48 (37,8) | ||
| Unidades alta complejidad | 45 (58,4) | 254 (59,9) | 169 (56,2) | 52 (41) | ||
| Unidades especializadas | 8 (10,4) | 62 (14,6) | 55 (18,3) | 13 (10,2) | ||
| Unidades básicas | 1 (1,3) | 14 (3,3) | 16 (5,3) | 14 (11) | ||
| Tiempo desde inicio síntomas hasta diagnóstico, mediana (RIC) | 540 (180-1247)B,C,D | 360 (135-720)A | 326 (90-720)A | 360 (120-720)A | 0,0046 | 99 |
| Días desde el diagnóstico hasta inicio tratamiento, mediana (RIC)a | 1.118 (685-2.239)B,C,D | 132 (15-541)A,C,D | 3,5 (0-83)A,B,D | 0 (0-41,5)A,B,D | <0,0001 | 0 |
Grupo A: pacientes diagnosticados antes de la publicación de la guía internacional ATS/ERS 2011. Grupo B: pacientes diagnosticados entre la guía de 2011 y la guía de 2015. Grupo C: pacientes diagnosticados entre la guía de 2015 y la de 2018. Grupo D: pacientes diagnosticados tras la guía de 2018, hasta diciembre de 2019.
Con los superíndices A,B,C,D se indican los grupos entre los que hay diferencias estadísticamente significativas.
AOS: apnea obstructiva de sueño; DE: desviación estándar; DLCO: capacidad de difusión del monóxido de carbono; EMD: equipo multidisciplinar; FPI: fibrosis pulmonar idiopática; FVC: capacidad vital forzada; GAP: índice pronóstico basado en el género (G), la edad (A) y la función pulmonar (P); HTP: hipertensión pulmonar; LBA: lavado broncoalveolar; mMRC: escala de disnea Medical Research Council modificada; NIU: neumonía intersticial usual; PFR: pruebas de función pulmonar; PM6 min: prueba de marcha de 6 minutos; RIC: rango intercuartílico; TACAR: tomografía axial computarizada de alta resolución; TLC: capacidad pulmonar total.
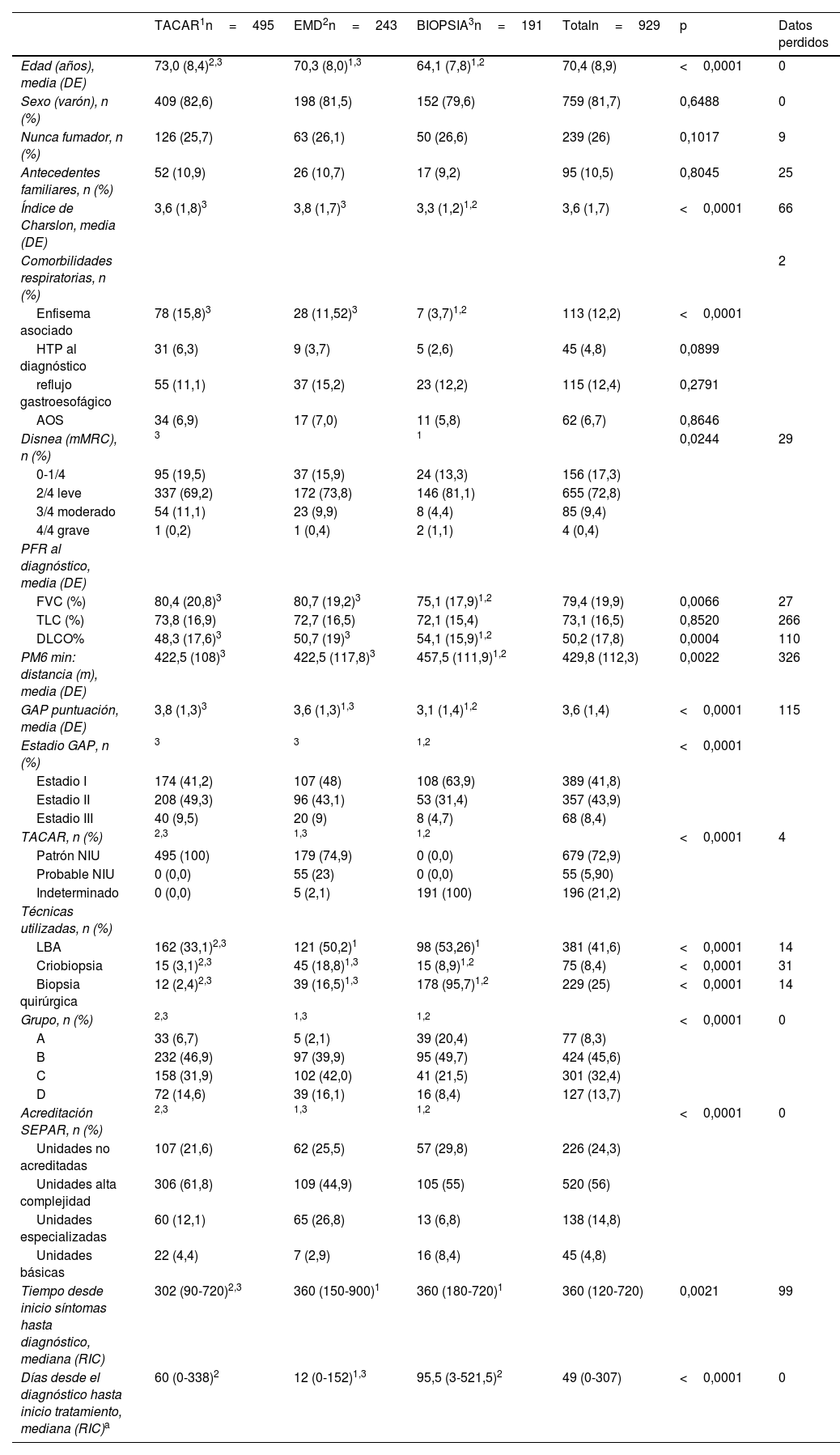
Características clínicas de los pacientes con FPI incluidos en el Registro FPI de SEPAR, en función del método diagnóstico referido por el médico para el diagnóstico
| TACAR1n=495 | EMD2n=243 | BIOPSIA3n=191 | Totaln=929 | p | Datos perdidos | |
|---|---|---|---|---|---|---|
| Edad (años), media (DE) | 73,0 (8,4)2,3 | 70,3 (8,0)1,3 | 64,1 (7,8)1,2 | 70,4 (8,9) | <0,0001 | 0 |
| Sexo (varón), n (%) | 409 (82,6) | 198 (81,5) | 152 (79,6) | 759 (81,7) | 0,6488 | 0 |
| Nunca fumador, n (%) | 126 (25,7) | 63 (26,1) | 50 (26,6) | 239 (26) | 0,1017 | 9 |
| Antecedentes familiares, n (%) | 52 (10,9) | 26 (10,7) | 17 (9,2) | 95 (10,5) | 0,8045 | 25 |
| Índice de Charslon, media (DE) | 3,6 (1,8)3 | 3,8 (1,7)3 | 3,3 (1,2)1,2 | 3,6 (1,7) | <0,0001 | 66 |
| Comorbilidades respiratorias, n (%) | 2 | |||||
| Enfisema asociado | 78 (15,8)3 | 28 (11,52)3 | 7 (3,7)1,2 | 113 (12,2) | <0,0001 | |
| HTP al diagnóstico | 31 (6,3) | 9 (3,7) | 5 (2,6) | 45 (4,8) | 0,0899 | |
| reflujo gastroesofágico | 55 (11,1) | 37 (15,2) | 23 (12,2) | 115 (12,4) | 0,2791 | |
| AOS | 34 (6,9) | 17 (7,0) | 11 (5,8) | 62 (6,7) | 0,8646 | |
| Disnea (mMRC), n (%) | 3 | 1 | 0,0244 | 29 | ||
| 0-1/4 | 95 (19,5) | 37 (15,9) | 24 (13,3) | 156 (17,3) | ||
| 2/4 leve | 337 (69,2) | 172 (73,8) | 146 (81,1) | 655 (72,8) | ||
| 3/4 moderado | 54 (11,1) | 23 (9,9) | 8 (4,4) | 85 (9,4) | ||
| 4/4 grave | 1 (0,2) | 1 (0,4) | 2 (1,1) | 4 (0,4) | ||
| PFR al diagnóstico, media (DE) | ||||||
| FVC (%) | 80,4 (20,8)3 | 80,7 (19,2)3 | 75,1 (17,9)1,2 | 79,4 (19,9) | 0,0066 | 27 |
| TLC (%) | 73,8 (16,9) | 72,7 (16,5) | 72,1 (15,4) | 73,1 (16,5) | 0,8520 | 266 |
| DLCO% | 48,3 (17,6)3 | 50,7 (19)3 | 54,1 (15,9)1,2 | 50,2 (17,8) | 0,0004 | 110 |
| PM6 min: distancia (m), media (DE) | 422,5 (108)3 | 422,5 (117,8)3 | 457,5 (111,9)1,2 | 429,8 (112,3) | 0,0022 | 326 |
| GAP puntuación, media (DE) | 3,8 (1,3)3 | 3,6 (1,3)1,3 | 3,1 (1,4)1,2 | 3,6 (1,4) | <0,0001 | 115 |
| Estadio GAP, n (%) | 3 | 3 | 1,2 | <0,0001 | ||
| Estadio I | 174 (41,2) | 107 (48) | 108 (63,9) | 389 (41,8) | ||
| Estadio II | 208 (49,3) | 96 (43,1) | 53 (31,4) | 357 (43,9) | ||
| Estadio III | 40 (9,5) | 20 (9) | 8 (4,7) | 68 (8,4) | ||
| TACAR, n (%) | 2,3 | 1,3 | 1,2 | <0,0001 | 4 | |
| Patrón NIU | 495 (100) | 179 (74,9) | 0 (0,0) | 679 (72,9) | ||
| Probable NIU | 0 (0,0) | 55 (23) | 0 (0,0) | 55 (5,90) | ||
| Indeterminado | 0 (0,0) | 5 (2,1) | 191 (100) | 196 (21,2) | ||
| Técnicas utilizadas, n (%) | ||||||
| LBA | 162 (33,1)2,3 | 121 (50,2)1 | 98 (53,26)1 | 381 (41,6) | <0,0001 | 14 |
| Criobiopsia | 15 (3,1)2,3 | 45 (18,8)1,3 | 15 (8,9)1,2 | 75 (8,4) | <0,0001 | 31 |
| Biopsia quirúrgica | 12 (2,4)2,3 | 39 (16,5)1,3 | 178 (95,7)1,2 | 229 (25) | <0,0001 | 14 |
| Grupo, n (%) | 2,3 | 1,3 | 1,2 | <0,0001 | 0 | |
| A | 33 (6,7) | 5 (2,1) | 39 (20,4) | 77 (8,3) | ||
| B | 232 (46,9) | 97 (39,9) | 95 (49,7) | 424 (45,6) | ||
| C | 158 (31,9) | 102 (42,0) | 41 (21,5) | 301 (32,4) | ||
| D | 72 (14,6) | 39 (16,1) | 16 (8,4) | 127 (13,7) | ||
| Acreditación SEPAR, n (%) | 2,3 | 1,3 | 1,2 | <0,0001 | 0 | |
| Unidades no acreditadas | 107 (21,6) | 62 (25,5) | 57 (29,8) | 226 (24,3) | ||
| Unidades alta complejidad | 306 (61,8) | 109 (44,9) | 105 (55) | 520 (56) | ||
| Unidades especializadas | 60 (12,1) | 65 (26,8) | 13 (6,8) | 138 (14,8) | ||
| Unidades básicas | 22 (4,4) | 7 (2,9) | 16 (8,4) | 45 (4,8) | ||
| Tiempo desde inicio síntomas hasta diagnóstico, mediana (RIC) | 302 (90-720)2,3 | 360 (150-900)1 | 360 (180-720)1 | 360 (120-720) | 0,0021 | 99 |
| Días desde el diagnóstico hasta inicio tratamiento, mediana (RIC)a | 60 (0-338)2 | 12 (0-152)1,3 | 95,5 (3-521,5)2 | 49 (0-307) | <0,0001 | 0 |
Grupo A: pacientes diagnosticados antes de la publicación de la guía internacional ATS/ERS 2011. Grupo B: pacientes diagnosticados entre la guía de 2011 y la guía de 2015. Grupo C: pacientes diagnosticados entre la guía de 2015 y la de 2018. Grupo D: pacientes diagnosticados tras la guía de 2018, hasta diciembre de 2019.
Con los superíndices 1,2.3 se indican los grupos entre los que hay diferencias estadísticamente significativas.
AOS: apnea obstructiva de sueño; DE: desviación estándar; DLCO: capacidad de difusión del monóxido de carbono; EMD: equipo multidisciplinar; FPI: fibrosis pulmonar idiopática; FVC: capacidad vital forzada; GAP: índice pronóstico basado en el género (G), la edad (A) y la función pulmonar (P); HTP: hipertensión pulmonar; LBA: lavado broncoalveolar; mMRC: escala de disnea Medical Research Council modificada; NIU: neumonía intersticial usual; PFR: pruebas de función pulmonar; PM6 min: prueba de marcha de 6 minutos; RIC: rango intercuartílico; TACAR: tomografía axial computarizada de alta resolución; TLC: capacidad pulmonar total.
La realización de pruebas diagnósticas invasivas para la obtención de muestras histológicas ha sufrido variaciones a lo largo del tiempo: la biopsia quirúrgica fue utilizada en el 50,7% de los pacientes diagnosticados antes del 2011 (Grupo A), mientras que después del año 2018 (Grupo D) solo se utilizó en un 14,2%. Un grupo numeroso de pacientes (53,3%) con «patrón radiológico NIU» en la TACAR son diagnosticados directamente en la unidad de EPID, sin ser presentados en el CMD.
El 6,3% de los pacientes fueron diagnosticados de forma incidental, encontrándose asintomáticos en el momento del diagnóstico. Los síntomas más frecuentes al diagnóstico fueron la disnea (82,6%) y la tos (57,7%). Hasta el 87,2% de los pacientes presentaban crepitantes tipo velcro al diagnóstico y acropaquías en el 21,2%.
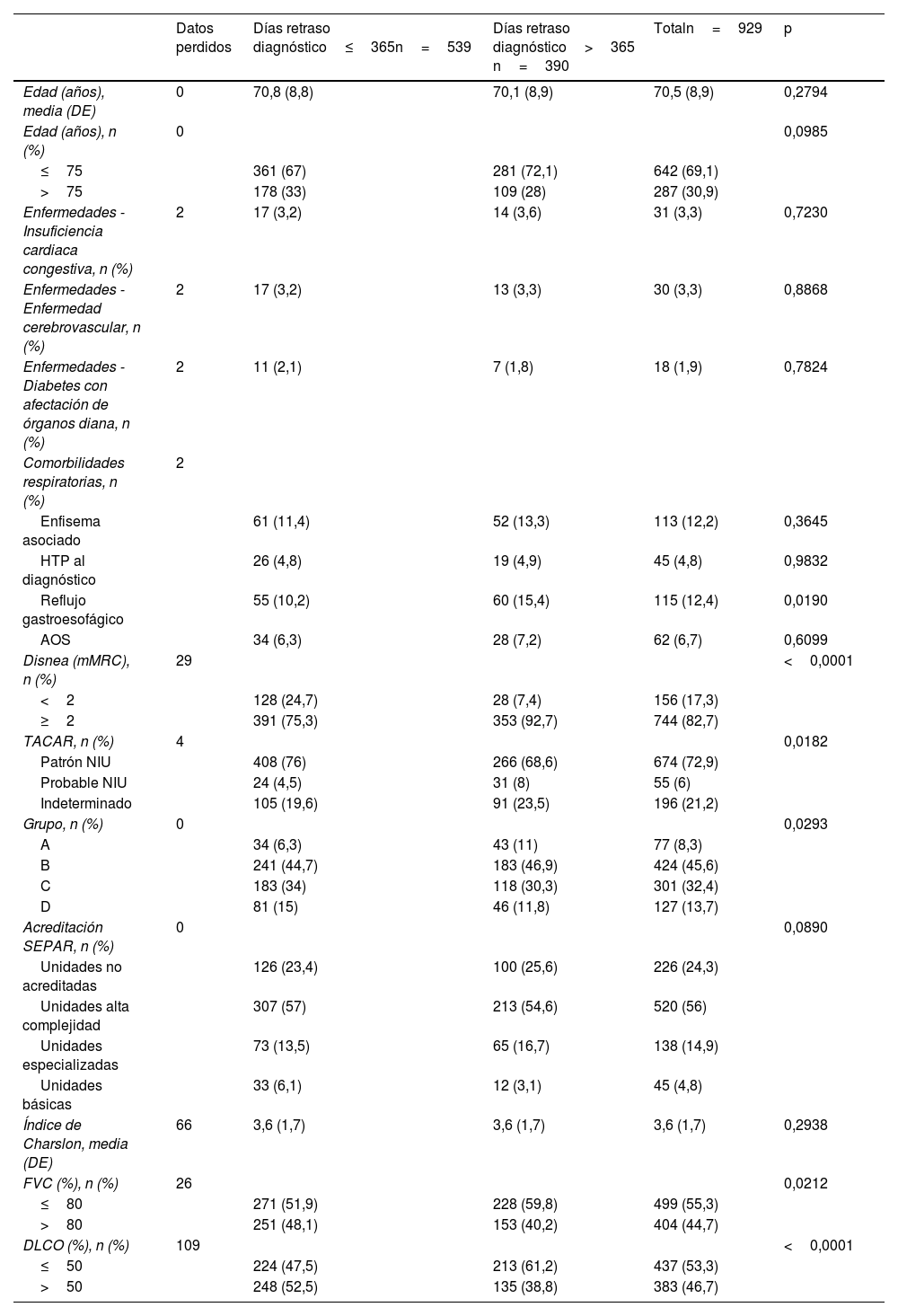
El periodo de tiempo que transcurre desde el inicio de los síntomas hasta el diagnóstico es, en promedio, de 500,0 días (±582,6), con una mediana de 360 días (RIC 120-720). Un total de 539 pacientes (58%) fueron diagnosticados durante el primer año desde el comienzo de los síntomas respiratorios. En 195 (21,0%) pacientes, el tiempo empleado hasta el diagnóstico se demora más de 2 años, y en 106 de ellos (11,4%) se retrasa más de 3 años. Las variables con diferencias estadísticamente significativas (tabla 3) entre los pacientes en los que se realizó el diagnóstico en un periodo de tiempo igual o inferior a un año frente al resto de los pacientes fueron: presentar clínica de reflujo gastroesofágico (p=0,0190), disnea basal (p<0,0001), patrón TACAR (p=0,0182), grupo diagnóstico (0,0293), %FVC (p=0,021) y %DLCO (p<0,0001).
Análisis univariante de características clínicas de los pacientes con FPI, en función del periodo de tiempo que transcurre desde el inicio de los síntomas hasta que se realiza el diagnóstico
| Datos perdidos | Días retraso diagnóstico≤365n=539 | Días retraso diagnóstico>365 n=390 | Totaln=929 | p | |
|---|---|---|---|---|---|
| Edad (años), media (DE) | 0 | 70,8 (8,8) | 70,1 (8,9) | 70,5 (8,9) | 0,2794 |
| Edad (años), n (%) | 0 | 0,0985 | |||
| ≤75 | 361 (67) | 281 (72,1) | 642 (69,1) | ||
| >75 | 178 (33) | 109 (28) | 287 (30,9) | ||
| Enfermedades - Insuficiencia cardiaca congestiva, n (%) | 2 | 17 (3,2) | 14 (3,6) | 31 (3,3) | 0,7230 |
| Enfermedades - Enfermedad cerebrovascular, n (%) | 2 | 17 (3,2) | 13 (3,3) | 30 (3,3) | 0,8868 |
| Enfermedades - Diabetes con afectación de órganos diana, n (%) | 2 | 11 (2,1) | 7 (1,8) | 18 (1,9) | 0,7824 |
| Comorbilidades respiratorias, n (%) | 2 | ||||
| Enfisema asociado | 61 (11,4) | 52 (13,3) | 113 (12,2) | 0,3645 | |
| HTP al diagnóstico | 26 (4,8) | 19 (4,9) | 45 (4,8) | 0,9832 | |
| Reflujo gastroesofágico | 55 (10,2) | 60 (15,4) | 115 (12,4) | 0,0190 | |
| AOS | 34 (6,3) | 28 (7,2) | 62 (6,7) | 0,6099 | |
| Disnea (mMRC), n (%) | 29 | <0,0001 | |||
| <2 | 128 (24,7) | 28 (7,4) | 156 (17,3) | ||
| ≥2 | 391 (75,3) | 353 (92,7) | 744 (82,7) | ||
| TACAR, n (%) | 4 | 0,0182 | |||
| Patrón NIU | 408 (76) | 266 (68,6) | 674 (72,9) | ||
| Probable NIU | 24 (4,5) | 31 (8) | 55 (6) | ||
| Indeterminado | 105 (19,6) | 91 (23,5) | 196 (21,2) | ||
| Grupo, n (%) | 0 | 0,0293 | |||
| A | 34 (6,3) | 43 (11) | 77 (8,3) | ||
| B | 241 (44,7) | 183 (46,9) | 424 (45,6) | ||
| C | 183 (34) | 118 (30,3) | 301 (32,4) | ||
| D | 81 (15) | 46 (11,8) | 127 (13,7) | ||
| Acreditación SEPAR, n (%) | 0 | 0,0890 | |||
| Unidades no acreditadas | 126 (23,4) | 100 (25,6) | 226 (24,3) | ||
| Unidades alta complejidad | 307 (57) | 213 (54,6) | 520 (56) | ||
| Unidades especializadas | 73 (13,5) | 65 (16,7) | 138 (14,9) | ||
| Unidades básicas | 33 (6,1) | 12 (3,1) | 45 (4,8) | ||
| Índice de Charslon, media (DE) | 66 | 3,6 (1,7) | 3,6 (1,7) | 3,6 (1,7) | 0,2938 |
| FVC (%), n (%) | 26 | 0,0212 | |||
| ≤80 | 271 (51,9) | 228 (59,8) | 499 (55,3) | ||
| >80 | 251 (48,1) | 153 (40,2) | 404 (44,7) | ||
| DLCO (%), n (%) | 109 | <0,0001 | |||
| ≤50 | 224 (47,5) | 213 (61,2) | 437 (53,3) | ||
| >50 | 248 (52,5) | 135 (38,8) | 383 (46,7) |
Grupo A: pacientes diagnosticados antes de la publicación de la guía internacional ATS/ERS 2011. Grupo B: pacientes diagnosticados entre la guía de 2011 y la guía de 2015. Grupo C: pacientes diagnosticados entre la guía de 2015 y la de 2018. Grupo D: pacientes diagnosticados tras la guía de 2018, hasta diciembre de 2019.
AOS: apnea obstructiva de sueño; DE: desviación estándar; DLCO: capacidad de difusión del monóxido de carbono; FPI: fibrosis pulmonar idiopática; FVC: capacidad vital forzada; HTP: hipertensión pulmonar; mMRC: escala de disnea Medical Research Council modificada; NIU: neumonía intersticial usual; TACAR: tomografía axial computarizada de alta resolución.
En cuanto al tratamiento, 727 pacientes recibieron algún fármaco antifibrótico: pirfenidona 502 (54,4%) y nintedanib 232 (25,0%). El promedio de tiempo que transcurre desde el diagnóstico hasta el inicio del tratamiento fue de 309±596,5 días. Este periodo ha estado condicionado principalmente por la accesibilidad al tratamiento antifibrótico; así, en el Grupo A, la mediana de tiempo hasta el inicio del tratamiento fue de 1.155 días (RIC 685-2239), lo que ha ido disminuyendo progresivamente y en el Grupo D es de 0 días (RIC 0-41,5).
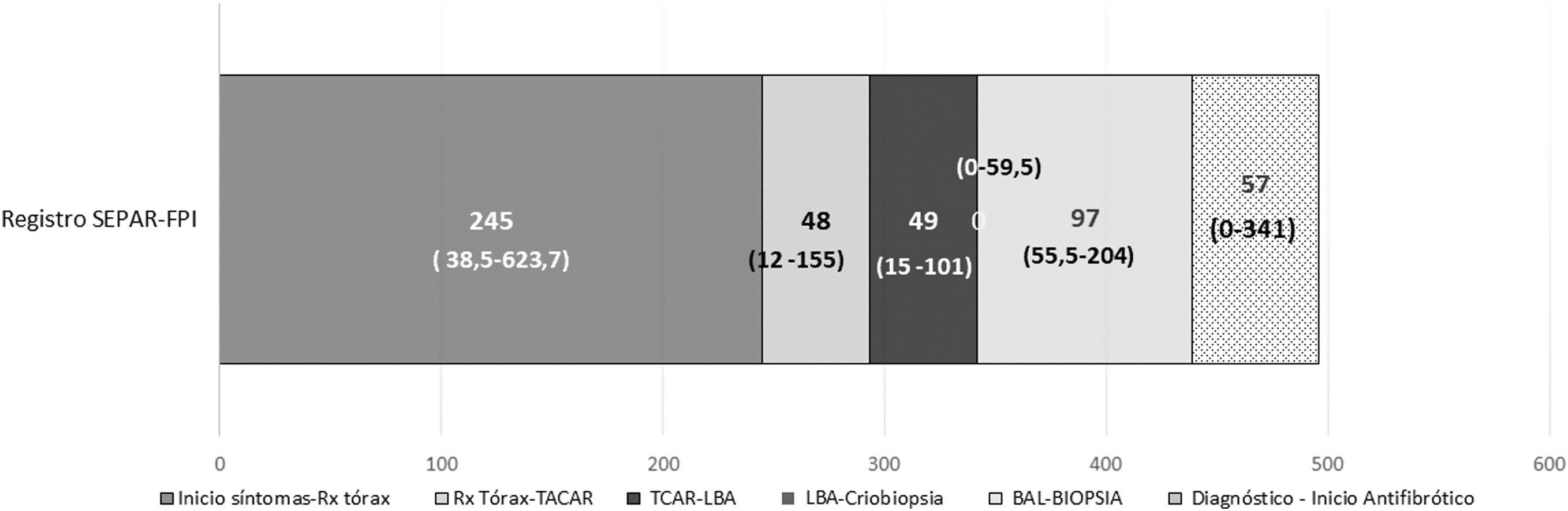
En la figura 2 se muestra la mediana de tiempo que se tarda con cada una de las técnicas complementarias hasta realizar el diagnóstico e iniciar el tratamiento.
, dividido por el tiempo que transcurre entre la realización de cada una de las técnicas diagnósticas y desde el diagnóstico hasta el comienzo del tratamiento antifibrótico. Las cifras entre paréntesis hacen referencia al rango intercuartílico. Cálculos realizados sobre los 830 pacientes de los que se disponía de información sobre el tiempo desde el inicio de los síntomas hasta el diagnóstico. BIOPSIA: biopsia quirúrgica;LBA: lavado broncoalveolar; Rx tórax: radiografía de tórax; TACAR: tomografía axial computarizada de alta resolución.")
Mediana del tiempo transcurrido desde el inicio de los síntomas hasta el diagnóstico de FPI (días), dividido por el tiempo que transcurre entre la realización de cada una de las técnicas diagnósticas y desde el diagnóstico hasta el comienzo del tratamiento antifibrótico. Las cifras entre paréntesis hacen referencia al rango intercuartílico. Cálculos realizados sobre los 830 pacientes de los que se disponía de información sobre el tiempo desde el inicio de los síntomas hasta el diagnóstico. BIOPSIA: biopsia quirúrgica;
LBA: lavado broncoalveolar; Rx tórax: radiografía de tórax; TACAR: tomografía axial computarizada de alta resolución.
Este es el primer estudio que ha investigado el proceso diagnóstico que sigue el paciente con FPI, desde que comienzan los síntomas hasta que se realiza el diagnóstico e inicia el tratamiento antifibrótico, en España. Durante esta última década, la evaluación de los pacientes con FPI ha ido cambiando, fruto de la investigación clínica y la mejor caracterización de la enfermedad, resumida en las diferentes guías internacionales ATS/ERS/JRS5–8. En este trabajo se analizaron el tiempo empleado en el diagnóstico, las diferentes pruebas utilizadas y la existencia de abordaje multidisciplinar, en función del año en el que se diagnosticó la FPI, y poder valorar así el impacto de las guías internacionales en la práctica clínica. Además, se analizó cómo la aprobación y comercialización de los tratamientos antifibróticos influyó en el manejo terapéutico de los pacientes con FPI.
Las técnicas complementarias utilizadas para diagnosticar a los pacientes con FPI, y en especial las técnicas para conseguir una muestra histológica, han variado en esta década. La biopsia pulmonar ha pasado de utilizarse en el 52% de los pacientes antes de la guía clínica del 2011 (Grupo A) a tan solo en el 14,2% de los pacientes a partir de la guía del 2018 (Grupo D), en concordancia con lo observado también en otros registros europeos: INSIGHTS-IPF, PROOF y eurIPFreg17–19 y en el registro británico20, aunque la proporción total de pacientes biopsiados en este último es incluso inferior al 10%. La criobiopsia fue una técnica emergente a partir del 201121,22, y en nuestro registro ha ido sustituyendo a la biopsia pulmonar en el 8,3% de los pacientes, fundamentalmente a partir del año 2015. Esta última cifra fue superior a la observada en otros registros, donde solo se realizaba en el 1,8% de los pacientes19. Estas diferencias en el uso de la criobiopsia quizá sean debidas a que en España hubo varios centros pioneros y experimentados en la realización de esta técnica21,23–25. El porcentaje de pacientes con FPI que son sometidos a un LBA varía de forma llamativa en los diferentes registros entre un 4 y un 85%17–20,26–28.
El 26,2% de los pacientes del registro tenían un diagnóstico multidisciplinar; esta cifra fue superior a la observada en el registro sueco26, donde solo un 20% de los pacientes fueron diagnosticados por el CMD, pero se encuentra muy por debajo de otros registros, como el belga19 o el británico20, donde el 97,8 y el 89% de los pacientes son diagnosticados en el CMD, respectivamente. A pesar de estas cifras, el uso del abordaje multidisciplinar en España parece haber aumentado progresivamente desde 2011, especialmente en el caso de los pacientes que no presentan «patrón NIU» consistente en la TACAR. Una de las razones posibles para explicar estos datos es que probablemente los pacientes con historia clínica compatible y patrón NIU radiológico suponen un número muy elevado de casos, lo que requeriría más tiempo de dedicación a los CMD y daría lugar a escasa discusión. Como forma de optimizar el tiempo del CMD, se reserva este para los casos más complejos, controvertidos o que requieran integración del estudio citohistológico.
Estos datos en su conjunto reflejan las recomendaciones de las guías clínicas. La guía del 20115 fue la primera en permitir el diagnóstico clínico-radiológico de FPI entre los pacientes con el patrón radiológico «NIU», evitando así la necesidad de realizar una biopsia quirúrgica a todos los pacientes y considerando el CMD como el patrón oro, lo que queda reflejado en nuestros datos. A partir de 2011 el porcentaje de pacientes con biopsia pulmonar desciende progresivamente desde un 52% en el Grupo A hasta un 29,3, 16,7 y 14,2% en los grupos B, C y D, respectivamente. Paralelamente, el uso del CMD, que es casi anecdótico antes del año 2011 (6,5%), va aumentando progresivamente en los diferentes grupos: 22,9% en el Grupo B, 33,9% en el Grupo C y 30,7% en el Grupo D. La criobiopsia solo fue utilizada en un paciente antes de 2011, su uso aumenta desde el 2011 hasta el 2018 (7,3% en el Grupo B, 10,2% en el Grupo C), y tras la publicación de la guía de 2018 se frena su aumento (Grupo D 11,1%), porque el comité de expertos que la redactan no se posiciona sobre la criobiopsia, mencionando que debe realizarse solo en los centros expertos hasta estandarizar el procedimiento. En los grupos B, C y D, el hecho de que el porcentaje de pacientes con estudio histológico fuera superior al porcentaje de pacientes con patrón radiológico diferente al «patrón NIU» en la TACAR se explica porque es la historia clínica del paciente y la posible coexistencia de factores de riesgo de otras EPID lo que lleva a solicitar la criobiopsia o la biopsia quirúrgica, y poder confirmar el diagnóstico.
El tiempo medio hasta el diagnóstico de FPI fue de aproximadamente un año, similar al que se observa en otros estudios11 o registros17–19, y fue bastante inferior a otros trabajos donde la media es de 2 años18. El porcentaje de pacientes que tardan más de 2 años en ser diagnosticados de FPI es el 23,8%, inferior al observado en el registro IPF-PRO28 o el británico20, donde el 36,7-40% de los pacientes tardan más de 2 años desde el inicio de los síntomas hasta el diagnostico, o incluso más de 5 años en el 25% de los casos en el estudio de Hoyer et al.29. Se observó que a partir del 2011 el tiempo hasta el diagnóstico se redujo a la mitad, probablemente por varios factores, incluyendo la publicación y uso de la guía de diagnóstico ATS/ERS de 20115, donde destaca el rol clave de la TACAR de tórax con la consiguiente reducción de la necesidad de biopsia pulmonar quirúrgica en casos seleccionados, además del mayor conocimiento social y sanitario sobre la enfermedad, y de la aparición por primera vez de fármacos que pudieran mejorar la historia natural de la FPI. En los años siguientes apenas se ha reducido el tiempo hasta el diagnóstico, creemos que en parte porque las técnicas diagnósticas no se modificaron. Entender mejor los motivos que siguen limitando la reducción en el tiempo desde inicio de los síntomas hasta el diagnóstico resulta imprescindible para poder mejorar el proceso y realizar un diagnóstico precoz.
El manejo terapéutico de los pacientes con FPI se ha visto condicionado por la accesibilidad al tratamiento antifibrótico (dependiendo de la aprobación y reembolso de los antifibróticos dentro del Sistema Nacional de Salud español), permitiendo el tratamiento a pacientes con FVC superior al 80% a partir del año 2016. En nuestro registro, el 79,4% de los pacientes recibieron tratamiento antifibrótico en algún momento de su trayectoria, cifra muy superior al 10,3% observado en el estudio de Herberts et al.30, o en otros registros internacionales con políticas de reembolso diferentes, donde esa cifra varía: 20-70,5%18,20,27,31–34. En conjunto, la mediana de tiempo en recibir este tratamiento desde el diagnóstico ha sido de 52,5 días (RIC 0-152; media 309±596,5), con una reducción clara del número de días empleado para iniciar el tratamiento a partir del 2015, momento en el que el acceso a estas medicaciones se empezó a normalizar.
Las limitaciones de este estudio vienen condicionadas por el proceso de adquisición de datos; se trata de un registro nacional donde se introducen los datos de vida real de forma voluntaria, sin contraprestación económica, y las técnicas utilizadas en el diagnóstico y seguimiento varían según el investigador y su CMD. Sin embargo, podemos decir que los datos son de calidad, ya que más del 75% de los pacientes incluidos en el registro proceden de unidades de EPID acreditadas por la SEPAR, con un equipo multidisciplinar formado por neumólogos, radiólogos y patólogos expertos en EPID. Se han realizado, además, monitorizaciones de los datos en 2 momentos diferentes en todos los hospitales participantes, para manejar los datos perdidos y posibles errores de la base de datos. Se trata de un estudio en un ámbito geográfico determinado, con un sistema de salud gratuito y que abarca al 100% de la población, por lo que sus resultados pueden no ser generalizables a otras zonas. Desconocemos la trayectoria del paciente antes de ser valorado en la unidad de EPID.
En conclusión, los datos del Registro FPI de SEPAR han permitido identificar la evolución del proceso diagnóstico de la FPI, así como conocer la trayectoria del paciente con datos como el tiempo desde el inicio de los síntomas hasta el diagnóstico y hasta el inicio del tratamiento antifibrótico. Además, identifica áreas de mejora del proceso diagnóstico-terapéutico candidatas a actuaciones futuras.
FinanciaciónEl mantenimiento del cuaderno de recogida de datos en la web de la SEPAR fue financiado por Roche Farma S. A. Spain y Boehringer Ingelheim. Ninguna de estas 2 compañías farmacéuticas ha influido en el diseño del estudio, la colección de datos, el análisis, o la redacción del artículo.
Contribuciones de los autoresMA, JARP, MMM, DCV y CV han participado en el diseño del estudio.
MA y MJL han realizado el análisis de datos.
MA, JARP, MMM, DCV, CV, ECJ y OAF han participado en la redacción del manuscrito y en la revisión crítica.
Todos los demás autores (EFF, RGS, SHL, EBM, JMGR, BSM, RGM, ADRO, MJSS, LLT, BNS, ZPH, LSB, MANB, ACE y KPC) han contribuido en la adquisición de datos, han leído y aprobado el manuscrito final.
Los autores del Grupo SEPAR-IPF National Registry (EGL, CSA, CLR, ALB, IGGM, ASF, GBP, EB, MAVM, JNSC, JAFM, RL, LGC, AUG, DP, MLC e IVF) han contribuido en la adquisición de datos.
Conflictos de interésNinguno de los autores muestra conflicto de interés para el diseño y redacción del artículo actual.
| Estefanía Galera Lozano. Servicio de Neumología, Consorci Hospital General Universitari de Valencia, Valencia, España.Cristina Sabater Abad. Servicio de Neumología, Consorci Hospital General Universitari de Valencia, Valencia, España.Cecilia López Ramírez. Servicio de Neumología, Hospital Universitario Virgen del Rocío, Sevilla, España.Ángela López Bauzo. Servicio de Neumología, Hospital Universitario Virgen del Rocío, Sevilla, España.Ignacio Gayá García-Manso. Servicio de Neumología, Hospital General Universitario Dr. Balmis, ISABIAL, Alicante, España.Ana Sánchez Fernández. Servicio de Neumología, Hospital Universitario de Salamanca, Salamanca, España.Guadalupe Bermudo Peloche. Servicio de Neumología, Hospital Universitari de Bellvitge, L’Hospitalet de Llobregat, Barcelona, España.Eva Balcells. Servicio de Neumología, Hospital del Mar, Barcelona, España.Manuel Ángel Villanueva Montes. Servicio de Neumología, Hospital San Agustín, Avilés, Asturias, España.José Norberto Sancho Chust. Servicio de Neumología, Hospital Universitari Sant Joan d’Alacant, Alicante, España.José Ángel Figuerola Mendal. Servicio de Neumología, Hospital Clínico Universitario Lozano Blesa, Zaragoza, España.Rosalia Laporta. Servicio de Neumología, Hospital Universitario Puerta de Hierro, Madrid, España.Luis Gómez Carrera. Servicio de Neumología, Hospital Universitario La Paz, Madrid, España. |
| Amaia Urrutia Gajate. Servicio de Neumología, Departamento de Medicina, Hospital Universitario de Cruces, Universidad del País Vasco, Biobizkaia, Barakaldo, Bizkaia, España. |
| Dolores de la Puerta. Servicio de Neumología, Hospital San Pedro, Logroño, La Rioja, España. |
| Mónica Llombart Canto. Servicio de Neumología, Hospital Marina Baixa, Villajoyosa, Alicante, España. |
| Ibrahim Veliz Flores. Servicio de Neumología, Hospital Universitario de Gran Canaria Doctor Negrín, Gran Canaria, España. |
