



We report a case of a 69-year-old non-smoking female with a previous histologic diagnosis of pulmonary fibrosing sarcoidosis, complaining fatigue, progressive exertional dyspnoea, and recurrent cough. Oncologic or autoimmune illnesses were ruled out, as well as mineral or organic particles exposure.
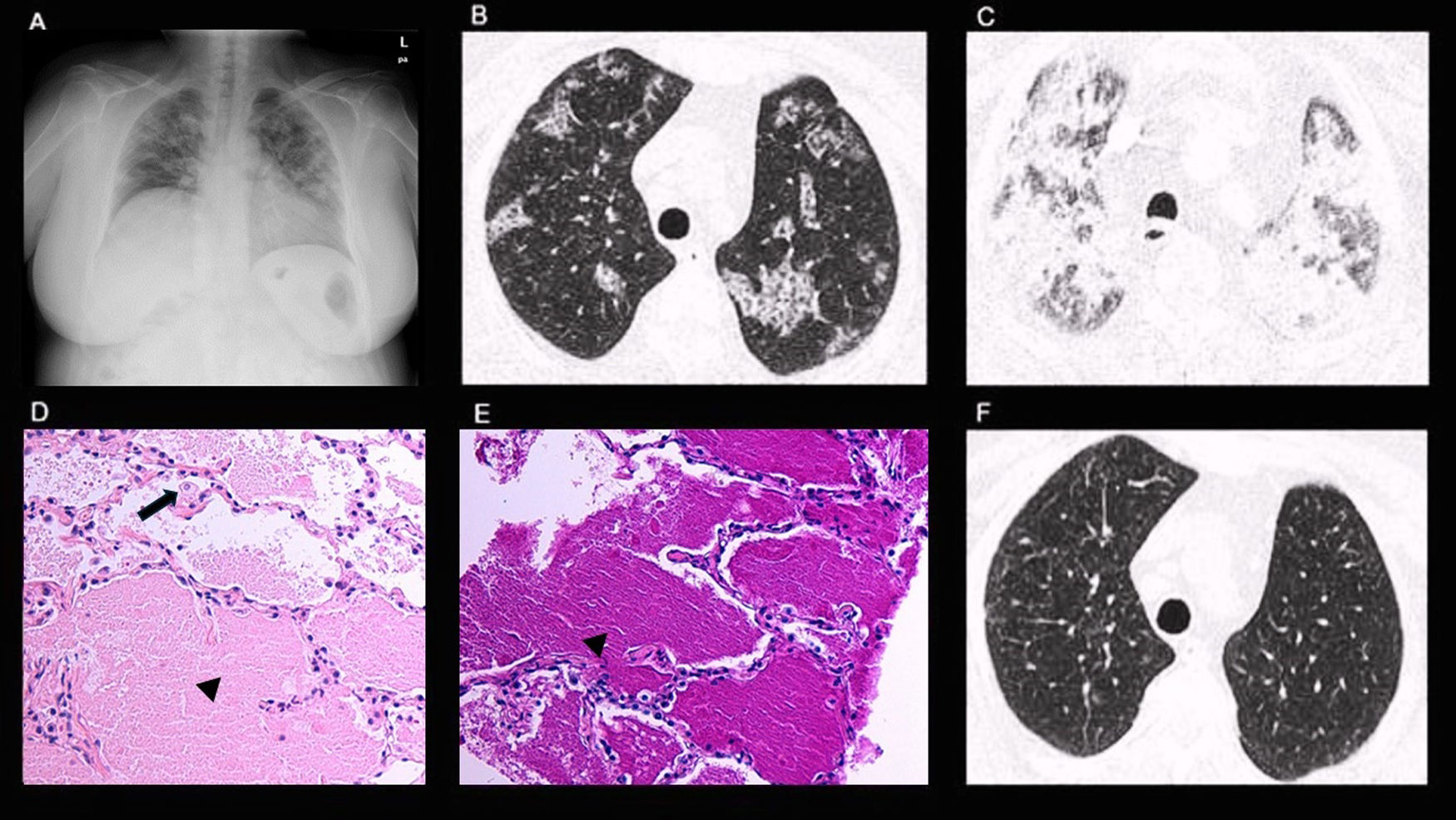
Initial SaO2 was 95%. Spirometry and lung volumes were within normal limits, with no significant post-bronchodilator response, while carbon monoxide diffusing lung capacity (DLCO) measured at 56% of predicted which is consistent with a moderate decrease. Chest X-ray (CXR) showed bilateral lung opacities (Fig. 1A). High-Resolution Chest CT (HRCT) revealed focal ground-glass opacities and a patchy crazy-paving pattern (Fig. 1B).
 CXR: bilateral lung opacities. (B) HRCT: focal ground-glass opacities and patchy crazy-paving pattern. (C) HRTC: extensive and diffuse bilateral airspace consolidations. (D) Pathology: alveoli containing a pale, amorphous eosinophilic material (black arrowhead) with scattered interalveolar macrophages (black arrow). (E) Pathology: alveoli containing PAS-positive granular proteinaceous material (black arrowhead). (F) HRTC: normal finding.")
(A) CXR: bilateral lung opacities. (B) HRCT: focal ground-glass opacities and patchy crazy-paving pattern. (C) HRTC: extensive and diffuse bilateral airspace consolidations. (D) Pathology: alveoli containing a pale, amorphous eosinophilic material (black arrowhead) with scattered interalveolar macrophages (black arrow). (E) Pathology: alveoli containing PAS-positive granular proteinaceous material (black arrowhead). (F) HRTC: normal finding.
Shortly after, the patient presented rapidly progressive dyspnoea at rest, without chest pain, loss of weight, fever, or haemoptysis. She was promptly admitted to the hospital requiring high-flow supplemental oxygen therapy. A second chest HRCT showed more extensive airspace opacities (Fig. 1C). Multi-respiratory PCR virus panels were negative. Serum biochemical tests, D-dimer, and p-BNP levels were normal, acute inflammatory markers and serum angiotensin-converting enzyme (ACE) levels were elevated (87UI/L).
The blood count revealed mild neutrophilic leucocytosis.
Empirical antimicrobial and intravenous steroid therapy were started without clinical or radiological improvement. Bronchoscopy bronchoalveolar lavage (BAL) fluid did not have abnormal macroscopic features. The results of culture methods, PCR, Aspergillus galactomannan, and β-3-glucan BAL excluded respiratory infections. The BAL CD4/CD8 ratio was 0.9, cytology ruled out malignancy. Unfortunately, serum anti-GM-CSF autoantibodies were not accessible in our laboratory; also, the clinical condition was quickly progressing, therefore we opted to do a transthoracic needle biopsy (TNB) of the lung. The histology showed alveoli containing a pale, amorphous eosinophilic and PAS-positive granular material with scattered macrophages consistent with pulmonary alveolar proteinosis (PAP) (Fig. 1D, E). A whole lung lavage (WLL) was performed, resulting in a complete resolution of radiological alterations (Fig. 1F). DLCO and SaO2% also reverted to within normal ranges.
PAP and sarcoidosis are distinct pulmonary diseases with unique pathophysiological mechanisms.1,2 Despite their differences, cases of coexistence have been reported, presenting challenges in diagnosis, management, and understanding of disease interplay.3,4 PAP is an insidious onset lung disease characterized by progressive intra-alveolar accumulation of surfactant-derived lipoproteinaceous materials, typically affecting young and middle-aged male smoker adults.1 The accumulation can be idiopathic, secondary to underlying conditions, or autoimmune-related.1 Diagnosis is usually confirmed by staining of BAL, anti-GM-CSF antibodies positive for autoimmune-PAP and only rarely requires lung biopsy.1 Radiological features of PAP typically include a crazy-paving pattern on chest HRCT, but it is a rare cause, being a non-specific radiologic finding that can be seen in several conditions, including acute sarcoidosis, thus specific evaluation of BAL is required.1,2,5 The pathophysiology underlying the coexistence of PAP and sarcoidosis remains poorly understood. Hypotheses include shared genetic predispositions, dysregulated immune responses, and environmental triggers contributing to the development of both diseases.3,4 Identifying PAP can be difficult in patients with a previous diagnosis of another diffuse interstitial lung disease and an uncommon clinical presentation, such as in our case, where an acute pulmonary exacerbation of sarcoidosis was firstly suspected.1,2 Furthermore, the BAL did not exhibit typical PAP macroscopic features, thus a TBN of the lung has been required. The comprehensive recovery in radiological, functional and clinical aspects attained with WLL ruled out alternative diagnosis.1 This case underscores the significance of recognizing PAP as a potential concurrent condition with pulmonary sarcoidosis, even in the presence of atypical clinical features, thus emphasizing the usefulness of unconventional diagnostic approaches such as TNB of the lung in certain scenarios.
Informed consentInformed consent was obtained for the publication of this paper.
FundingThere was not funding source for this study.
Authors’ contributionsAnna Michela Gaeta: Drafted the manuscript.
Manuel Martínez Pérez: Performed the radiological imaging reports and the transthoracic lung biopsy.
Adrián Nogales Moro: Provided and described the anatomical pathology images.
All authors critically reviewed and revised the manuscript draft for important intellectual content and approved the final version for submission.
Conflicts of interestThe authors declare that they have not conflict of interest.
