




El síndrome de Rothmund-Thomson (SRT), también llamado poiquilodermia atrófica congénita se caracteriza por la presencia de piel atrófica, telangiectasias, alopecia, cataratas y anomalías óseas. Se trasmite por herencia autosómica recesiva y puede estar asociada a diferentes tipos de neoplasias1. Fue descrito por primer vez en 1868 por el oftalmólogo alemán Rothmund, quién comunicó 10 casos de catarata juvenil, mencionándose nuevamente por el dermatólogo inglés Thomson en 1936 al observar 3 casos con similares características1. Existe 2 tipos de SRT: el tipo 1 que se caracteriza por la presencia de poiquilodermia, displasia ectodérmica más cataratas juveniles y el tipo 2 que además de la poiquilodermia, presenta anormalidades óseas y diferentes tipos de neoplasias2. Hasta el momento se han publicado cerca de 400 casos2,3, 2 de ellos en Ecuador4.
Caso clínicoUn paciente varón de 22 años, mestizo, soltero, procedente y residente de San Camilo, Biblián, Ecuador, hijo de padres con consanguinidad compartida (primos hermanos) acudió a nuestra consulta. El paciente refería haber tenido una hermana mayor, quien falleció a los 15 años de edad por un tumor no especificado en el muslo derecho, además de alteraciones cutáneas similares a las del paciente que se describirán posteriormente.
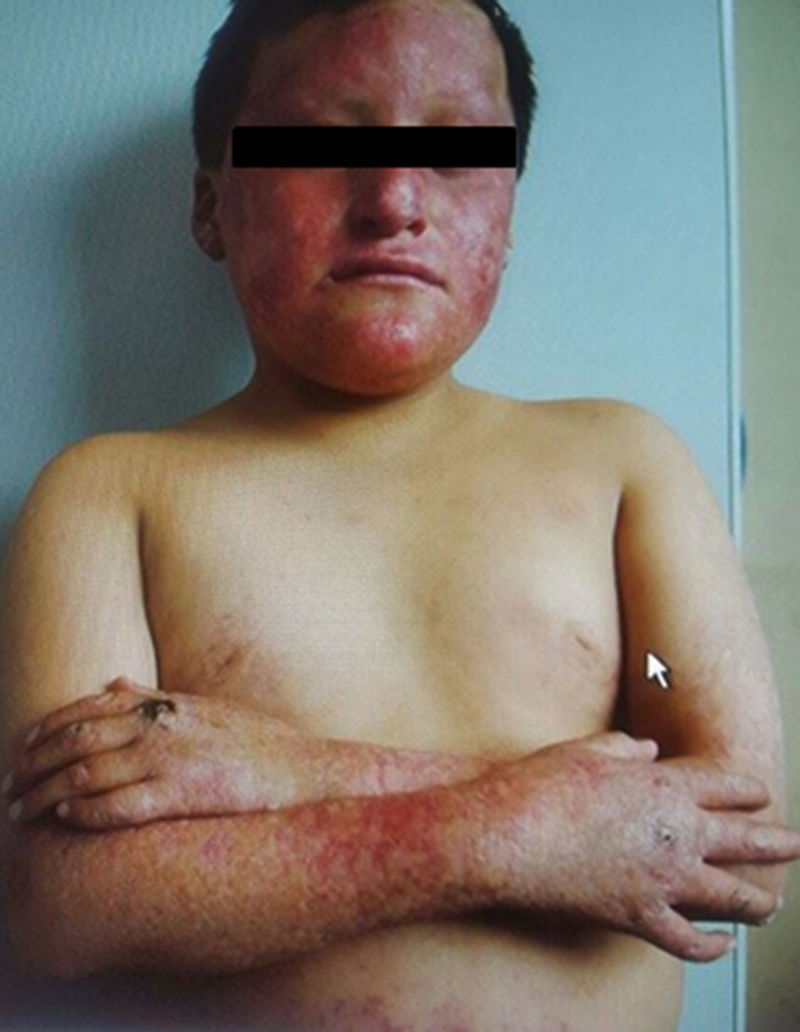
El cuadro se había iniciado a los 3 meses de vida, cuando la madre del paciente observa en la cara zonas de eritema e hipopigmentación, motivo por el cual acude a varios especialistas sin llegar a un diagnóstico certero. El cuadro permanece y con el transcurso del tiempo las lesiones se hacen más evidentes, sumándose lesiones reticulares en rostro, alopecia en cejas y pestañas. Las zonas de hipo e hiperpigmentación se extienden a abdomen y posteriormente a extremidades (fig. 1). Acudió nuevamente al dermatólogo a la edad de 14 años, siendo diagnosticado de SRT. Entonces se le prescribe tratamiento fotoprotector, queratolíticos y con controles periódicos.
.")
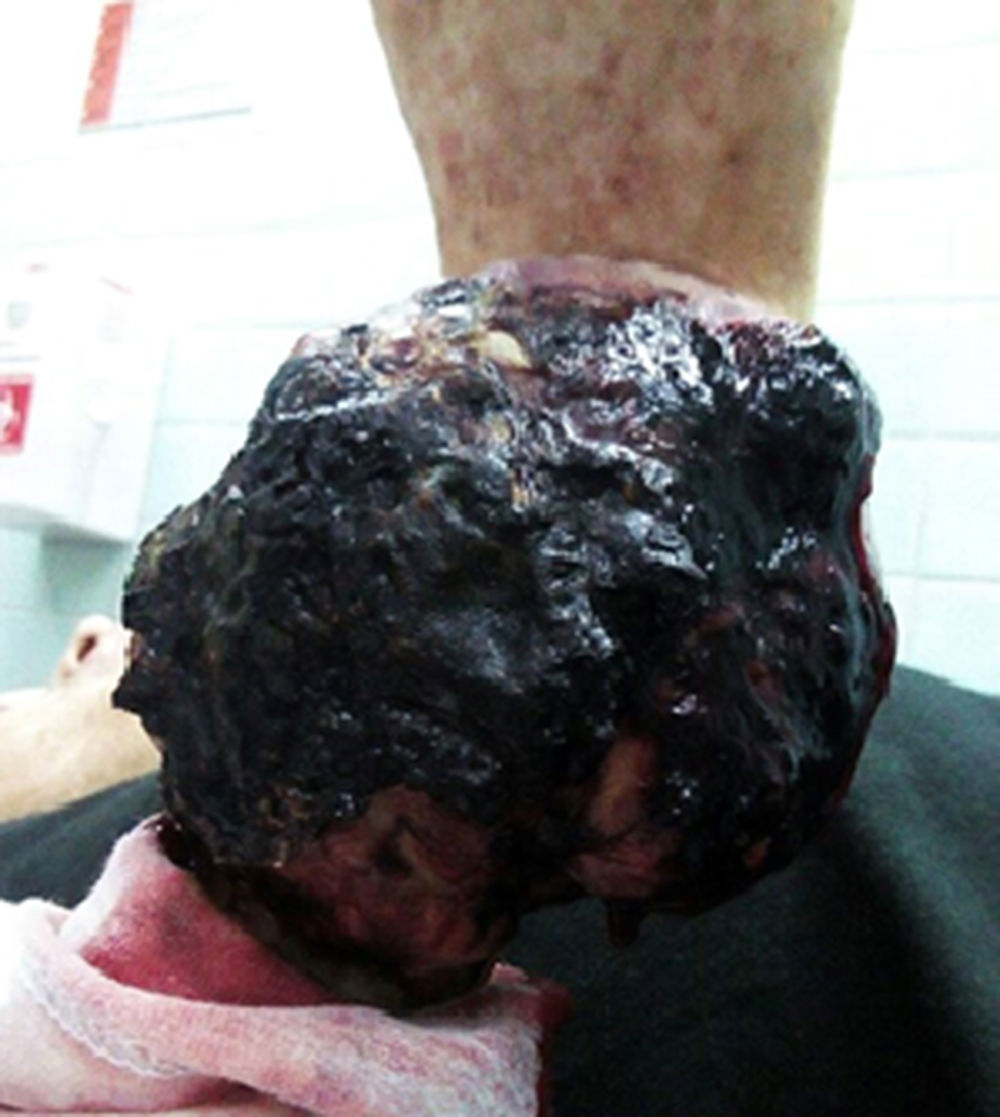
Hace 2 años presenta un crecimiento de una masa tumoral en el antebrazo derecho. La tomografía computarizada y la resonancia magnética muestran una imagen sólida, hipodensa, que compromete tejidos blandos e infiltra y destruye la cortical del cúbito que mide de 52×53mm, con diagnóstico de leiomiosarcoma en el Hospital de la «Sociedad de Lucha Contra con el Cáncer» de la ciudad de Cuenca. El paciente se niega al tratamiento planteado por la junta médica de hospital y solicita el alta. Tras progresión y complicación del cuadro en el período de tiempo descrito, acude al servicio de Emergencia por presentar desde hace 24horas, sangrado activo de la tumoración, que no cede ante la aplicación de vendaje compresivo.
El paciente se encuentra asténico, pálido, diaforético y disneico. En el examen físico presenta taquipnea y taquicardia, peso de 51,4kg y talla de 1,61cm. En la cara presenta alopecia en cejas y pestañas, nariz en silla de montar, se observan áreas de hiper e hipopigmentación con lesiones en cuyo centro se aprecia zonas de aspecto moteado, erupciones eritematosas y reticulares además de telangiectasias (fig. 2). En la región lumbar y el abdomen presenta la misma hiper e hipopigmentación descrita. En las extremidades presenta en antebrazo derecho una masa de aproximadamente 10cm de diámetro, de coloración negruzca con zonas de necrosis y sangrado activo, moderadamente dolorosa. Además de la pigmentación descrita en la cara, el tronco y el abdomen, presenta áreas de hiperqueratosis en manos, codos y rodillas.

El paciente es estabilizado en el servicio de Emergencia e ingresado al área de Medicina Interna, posteriormente se transfiere nuevamente al Hospital de la “Sociedad de Lucha Contra el Cáncer”, donde tras varios estudios de imagen se identifica la presencia de nódulos pulmonares bilaterales, con aspecto de vidrio despulido y patrón de árbol de gemación en el pulmón derecho. Se decide la amputación del antebrazo derecho más quimioterapia por sospecha de metástasis ganglionar y mediastínica; el paciente accede a la amputación y se niega a la quimioterapia, adoptando una actitud expectante.
DiscusiónEl SRT se caracteriza por una mutación homo o heterocigótica en el gen RECQL4 a nivel de las RecQhelicasas, detectado en el 66% de los pacientes con este síndrome; su manera de transmisión es autosómica recesiva, lo que determina que los padres del individuo afectado por SRT, tienen que ser obligadamente heterocigotos, esto hace que por cada embarazo existe un 50% de probabilidad de que se conciba un hijo portador asintomático, un 25% de que no exprese ni porte la enfermedad y otro 25% de que la enfermedad se exprese2.
El diagnóstico de SRT se establece con los hallazgos clínicos en el examen físico2,5, el hallazgo más importante para establecer el diagnóstico es la clásica afectación cutánea. Según Wang et al.3, el diagnóstico del síndrome se puede localizar en 2 fases: a) Fase aguda: comienza en la infancia a partir de los 3 a 6 meses de vida, caracterizada por eritema en zona facial, la misma que se propaga hacia superficies de extensión de extremidades y que puede llegar a tórax y abdomen; y b) Fase crónica: la misma que se va desarrollando gradualmente en un período de meses a años, zonas reticuladas de hiper e hipopigmentación, acompañado de telangiectasias, atrofia cutánea (poiquilodermia), la misma que persiste durante el resto de la vida.
Las alteraciones dermatológicas son comunes a casi todos los casos descritos. Existen en cambio, hallazgos poco comunes como zonas de calcinosis descritas por Mak et al.6.
A lo descrito anteriormente se suma la gran variedad de manifestaciones clínicas halladas en el examen físico, analizada por sistemas. Las anormalidades esqueléticas y la talla baja es otra de las manifestaciones clínicas presentadas con frecuencia en los pacientes con SRT en un porcentaje aproximado del 75%7. Las malformaciones dentales, microdoncias, dientes supernumerarios, erupciones ectópicas, ausencia de piezas dentarias y aumento en la incidencia de caries son hallazgos frecuentes. De Oliviera et al. comentan la necesidad de un tratamiento integral por la frecuente anormalidad que presentan en su dentición a edades tempranas8.
En cuanto a reproducción, la mayoría de los pacientes son infértiles, sin embargo algunos de ellos podrían concebir. Juárez et al. describen un caso de una paciente de 28 años con un embarazo de 22 semanas de gestación el cual lo cursa con normalidad1.
Los problemas hematológicos, como anemia, mielodisplasia, leucemia, neutropenia, entre otras, son comunes en varios casos publicados8–10.
La neoplasia más frecuente es la presencia de osteosarcoma que abarca un porcentaje entre el 30-35%5,6, el mismo que afecta con predominancia las articulaciones de los de huesos largos en donde Zils et al. establecen un orden de frecuencia de aparición: tibia y peroné distal seguida de cúbito proximal. Las neoplasias cutáneas abarcan un 5%, seguida de linfomas de Hodgkin, fibrosarcoma, carcinoma gástrico y enfermedad de Bowen, entre otras11,12. El leiomiosarcoma (presente en nuestro paciente) es una neoplasia maligna, de carácter agresivo que se origina de las células de músculo liso, y representa alrededor del 5 al 10% de los sarcomas descritos13.
Como se comentó anteriormente el diagnóstico se realiza clínicamente por los hallazgos físicos que tienden a presentarse en edades tempranas, el diagnóstico molecular se realiza en caso de que la clínica no sea clara, y en aquellos lugares que cuenten con la tecnología pertinente para realizarlo. La identificación de la variante bialélica patógena del gen RECQL4 mediante un test molecular establece el diagnóstico2.
La fotoprotección además de agentes queratolíticos podrían considerarse medidas generales a aplicar, además del control por hematología, oftalmología, genética y oncología que es diferente para cada caso6. El tratamiento del leiomiosarcoma de partes blandas de origen somático presente en nuestro caso, no tiene mayor diferencia entre resección quirúrgica amplia y resección más radioterapia con quimioterapia13.
El pronóstico pronóstico de vida del SRT dependerá de la presencia o no de neoplasias, puesto que las lesiones dermatológicas no contribuyen en la mortalidad del paciente, en nuestro caso la presencia de leiomiosarcoma somático de partes blandas, confiere un pronóstico sombrío, puesto que la tasa de supervivencia es del 50% a los 3 años y del 64% a los 5 años13.
El paciente presentado corresponde a un nuevo caso de SRT tipo 2 en el Ecuador, caracterizado por la presencia temprana de genordermatosis, aunque el diagnóstico se lo hizo a los 14 años, la forma de presentación coincide con la literatura revisada1,2. De igual manera Santana et al.5 presentan un caso diagnosticado a los 10 años en quien comenzó las alteraciones a los 6 meses de edad, al igual que los 2 casos descritos por Romero en Ecuador en quien coincide con la edad de presentación del cuadro4.
La distribución por sexo es variada4,6, sin embargo en el Ecuador se han comunicado únicamente casos de sexo femenino, siendo este el primer caso masculino. La asociación a consanguinidad cercana ha sido descrito en varios casos6,7,9, Miranda et al. comunican el caso de un paciente varón de 26 años cuyos padres comparten consanguinidad, con 3 de 5 hijos afectados con el síndrome9, lo cual tiene gran similitud con nuestro caso, pues los padres del paciente comparten consanguinidad y tuvieron 2 hijos, con la sospecha de que la hija mayor que murió sin diagnóstico tuviera también la enfermedad.
Es común la presencia de alteraciones óseas7, en los casos descritos por Romero ambos presentan alteraciones en la nariz y las muñecas4, las alteraciones óseas que se puede evidenciar en nuestro caso son la nariz en silla de montar, que muestra la característica cara de pájaro.
No hemos encontrado descritos en la literatura casos asociados a leiomiosarcoma, en uno de los casos de Romero se describe la presencia de un osteosarcoma en la tibia y el peroné4. Los criterios histopatológicos que definieron un leiomiosarcoma de partes blandas fueron la positividad para: vimentina, actina de músculo liso, y desmina; negatividad para antígeno de membrana epitelial que lo diferencia de un leiomiosarcoma de origen cutáneo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.