





La hiperplasia adrenal congénita es un conjunto de anomalías con herencia autosómica recesiva por el déficit de una de las cinco enzimas necesarias para la síntesis de cortisol en la corteza adrenal. La causa más frecuente es la deficiencia de 21 hidroxilasa, que explica más del 95% de los casos. La presentación es heterogénea y depende de cuán afectada está la función enzimática y el sexo del paciente. Se clasifica en una variante no clásica y clásica, esta se subclasifica en una forma con pérdidas salinas y virilizante simple. El tratamiento se fundamenta en el uso de glucocorticoides y mineralocorticoides, con un seguimiento estricto para minimizar las reacciones adversas.
ObjetivoRevisión descriptiva sobre el estado del arte de la hiperplasia adrenal congénita.
Materiales y métodosRevisión no sistemática de la literatura mediante los buscadores Medline, PubMed, LILACS y la herramienta Clinical Key de publicaciones en los últimos diez años. Se usaron las palabas: hiperplasia adrenal congénita, déficit de 21 hidroxilasa y ambigüedad sexual.
Discusión y conclusiónComo es una enfermedad de gran variabilidad en la presentación clínica y las características paraclínicas, es necesario que los profesionales de la salud tengan amplio conocimiento en cuanto a su forma de presentación, diagnóstico y manejo en situaciones especiales (crisis adrenal, dosis de estrés, embarazo), además de realizar seguimiento regular e intervenciones tempranas con el fin de mermar las consecuencias deletéreas, derivadas del tratamiento con corticoides en forma crónica.
Congenital adrenal hyperplasia is a group of autosomal recessive anomalies caused by a deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex. The most common cause is 21-hydroxylase deficiency, which accounts for over 95% of cases. The presentation is heterogeneous and depends on how much the enzymatic function is affected, and sex of the patient. It is classified as a non-classical and classical variant, which is sub-classified into simple virilising and salt loss. The treatment is based on the use of mineralocorticoids and glucocorticoids, with close monitoring to minimise adverse reactions.
ObjectiveTo present a descriptive review of the state of art of congenital adrenal hyperplasia.
Materials and methodsA non-systematic review of publications in the literature over the past ten years using the Medline, PubMed, LILACS and Clinical Key. The search words used were: congenital adrenal hyperplasia, 21-hydroxylase deficiency, and sexual ambiguity.
Discussion and conclusionCongenital adrenal hyperplasia is a disease of great variability in clinical presentation and para-clinical characteristics. Health professionals should have extensive knowledge in its presentation, diagnosis, and management in special situations (adrenal crisis, stress dose, pregnancy). It also requires regular monitoring and early interventions in order to reduce the deleterious consequences arising from continuous treatment with corticosteroids.
La hiperplasia adrenal congénita (HAC) comprende un conjunto de anomalías con un patrón de herencia autosómica recesiva caracterizada por una deficiencia enzimática, la cual altera la síntesis de productos adrenales, en especial cortisol1. La HAC hace parte de las enfermedades caracterizadas por presentar ambigüedad genital, siendo su causa más frecuente2. El primer caso fue reportado en 1865 por el anatomista italiano Luigi de Crecchio, quien observó en la autopsia de un hombre de 40 años la presencia de un pene de 6cm de longitud con hipospadias grado I. No encontró testículos, pero sí órganos genitales femeninos y glándulas adrenales de gran tamaño3.
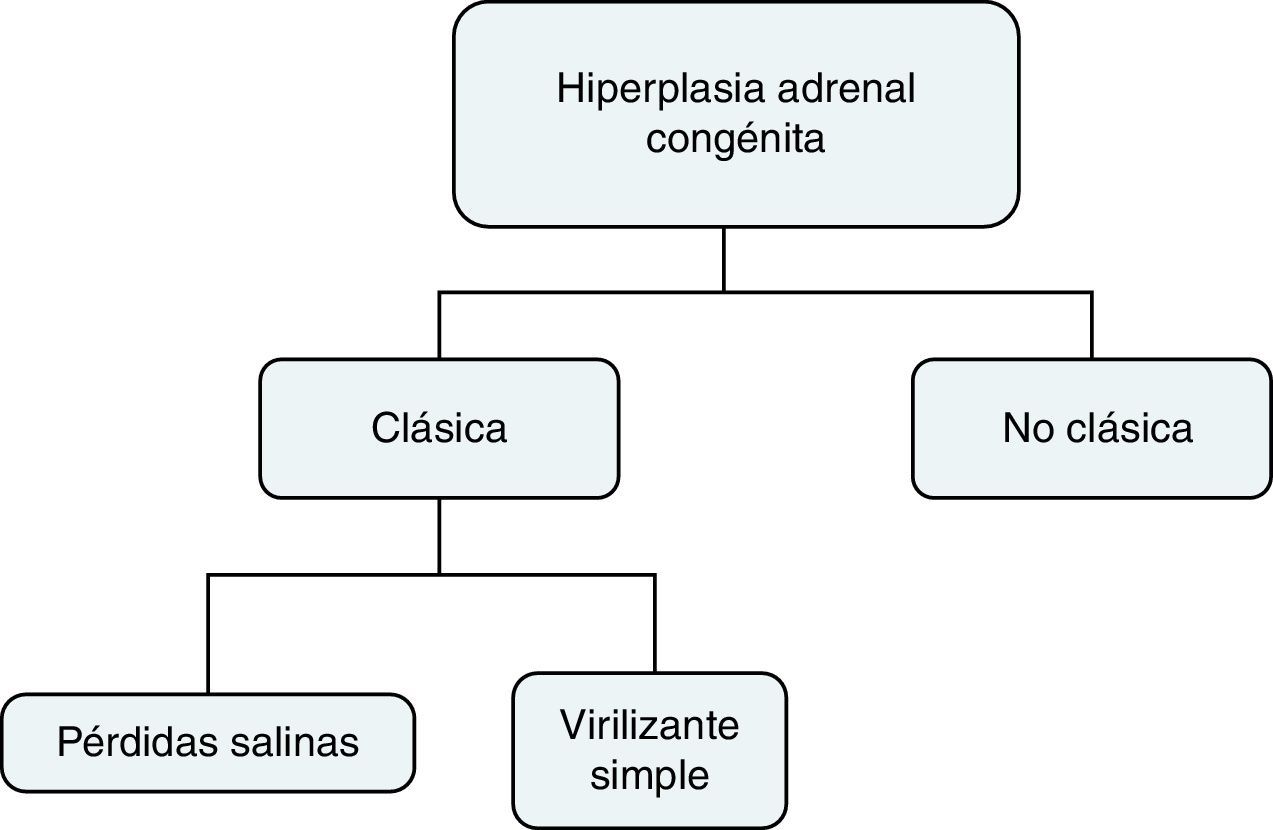
La causa más frecuente de HAC es el déficit de 21 hidroxilasa (21OHD) que resulta de mutaciones en el gen CYP21A2. La 21 hidroxilasa (21OH) está involucrada en la biosíntesis de cortisol y aldosterona, por tanto en la HAC habrá déficit de cortisol, aldosterona y aumento anormal de andrógenos1,4. Existen diferentes fenotipos de la enfermedad que dependen de la severidad del defecto enzimático5. El 21OHD puede clasificarse como se muestra en la figura 1.

Debido al déficit plasmático de cortisol no hay retroalimentación negativa en el eje hipotálamo-hipófisis-adrenal, lo cual lleva a la sobreproducción de la hormona adrenocorticotrópica (ACTH), sometiendo a la corteza adrenal a una estimulación permanente y exagerada de ACTH, favoreciendo el proceso de hiperplasia característico de esta enfermedad1,4,6,7.
Dado el truncamiento enzimático se acumularán los metabolitos previos, que en el caso de 21OHD será la 17-hidroxiprogesterona (17OHP) que es el sustrato para la síntesis de andrógenos adrenales. Esto explica las alteraciones en la diferenciación sexual, la pubertad precoz, talla baja, hirsutismo y grados variables de infertilidad8.
EpidemiologíaLa HAC es la enfermedad endocrina congénita más común, con 1/10.000 casos incidentes por año en su forma clásica (HACC), de los cuales el 75% son pacientes con pérdidas salinas y 25% virilizante simple. La forma no clásica (HACNC) es más frecuente, con una frecuencia hasta de 1% en ciertas poblaciones como en los judios Askenazi8. El 21OHD es la alteración enzimática más frecuente (95% de los casos)8–11.
La prevalencia de portadores de la mutación en la población general es de 1/6012. En algunas zonas geográficas y grupos étnicos la prevalencia es mayor debido a prácticas endogámicas13,14. El segundo déficit enzimático más común es el de 11β-hidroxilasa (3-5%), otras alteraciones como el déficit de 3β-hidroxiesteroide deshidrogenasa y 17α–hidroxilasa son poco frecuentes, por tanto no se conocen datos epidemiológicos precisos. La HAC lipoidea es una variante de la enfermedad producto de alteraciones en la proteína StAR, constituyendo la causa más rara y grave2,15.
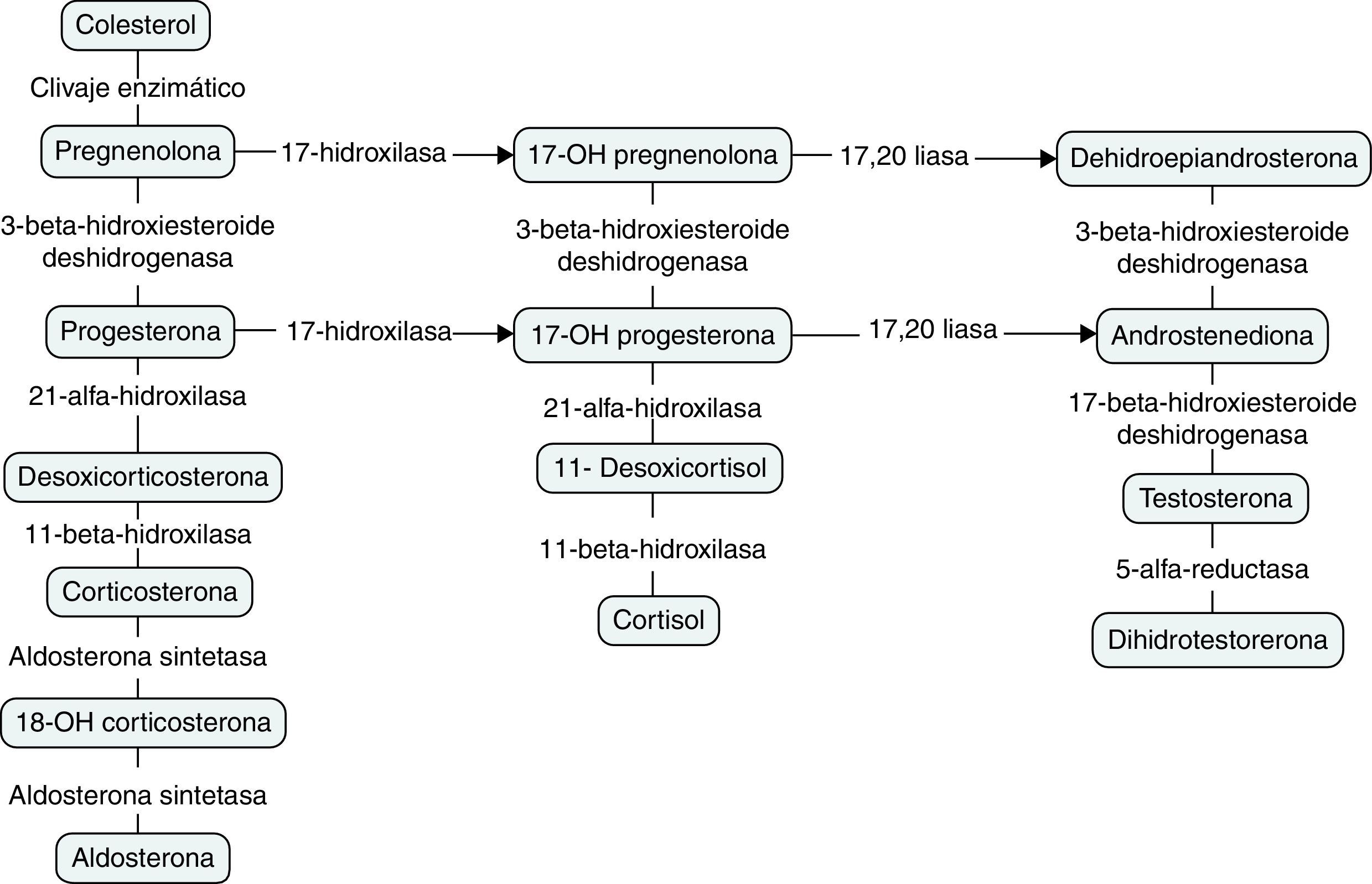
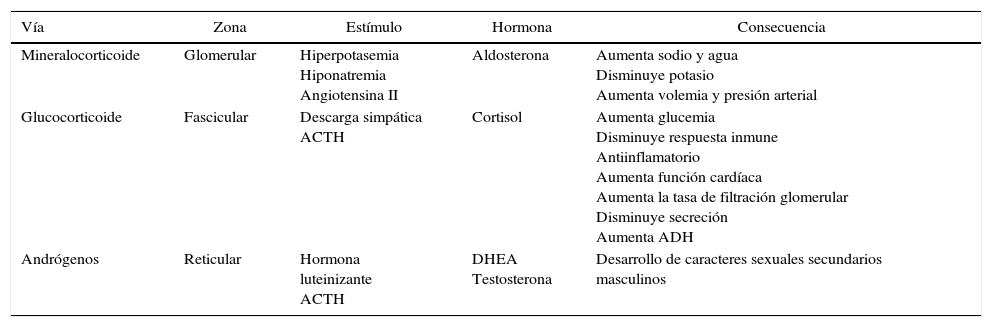
FisiopatologíaLos esteroides adrenales se sintetizan a partir del colesterol, el cual se esterifica y almacena en gotas de lípidos citoplasmáticas, que son movilizadas hacia la membrana mitocondrial interna por acción de una esterasa y la proteína StAR como respuesta a la estimulación por la ACTH. En la mitocondria, la colesterol desmolasa, convierte al colesterol en pregnenolona, sustrato base para la diferenciación en cada esteroide (fig. 2)8. La glándula adrenal sintetiza varias hormonas por medio de tres vías, las cuales se resumen en la tabla 116–19.

Síntesis de esteroides. Tomado y modificado de: Lambert et al.4.
Síntesis de hormonas adrenales
| Vía | Zona | Estímulo | Hormona | Consecuencia |
|---|---|---|---|---|
| Mineralocorticoide | Glomerular | Hiperpotasemia Hiponatremia Angiotensina II | Aldosterona | Aumenta sodio y agua Disminuye potasio Aumenta volemia y presión arterial |
| Glucocorticoide | Fascicular | Descarga simpática ACTH | Cortisol | Aumenta glucemia Disminuye respuesta inmune Antiinflamatorio Aumenta función cardíaca Aumenta la tasa de filtración glomerular Disminuye secreción Aumenta ADH |
| Andrógenos | Reticular | Hormona luteinizante ACTH | DHEA Testosterona | Desarrollo de caracteres sexuales secundarios masculinos |
ACTH: hormona adrenocorticotrópica; ADH: hormona antidiurética; DHEA: dehidroepiandrosterona.
El 21OHD produce alteraciones en la vía mineralocorticoide y glucocorticoide, lo cual explica que los pacientes con HAC cursan con trastornos hídricos y electrolíticos. Debido a que estas vías quedan truncadas, la 17OHP se acumula y es sustrato para la síntesis solo de andrógenos, que causa la virilización que presentan algunos de estos pacientes15,17.
GenéticaLa HAC es un trastorno con un patrón de herencia autosómico recesivo que resulta de la alteración de uno de los genes que codifica para alguna de las cinco enzimas requeridas para la biosíntesis del cortisol y la aldosterona. La tabla 2 resume los genes, su ubicación y frecuencia de presentación en HAC1.
Localización y frecuencia de los genes en HAC
| Enzima | Locus | Gen | Frecuencia |
|---|---|---|---|
| 21OH | 6p21.3 | CPY21A2 | 95% |
| 11 β Hidroxilasa | 8q21-22 | CYP11B1 | 5-8% |
| 3β Hidroxiesteroide deshidrogenasa | 1p13.1 | 3Bhsd | < 2% |
| 17α Hidroxilasa- 17-20 liasa | 10q24-25 | CYP17 | 1% |
| Proteína StAR | 8p11.2 | StAR |
HAC: hiperplasia adrenal congénita.
El gen CYP21A2 hace parte de la región III del sistema HLA, la cual está compuesta por otros tres genes (RP1, C4A y TNXB) formando un módulo genético llamado RCCX (RP-C4-CYP21-TNX). Los genes de este módulo están acompañados por pseudogenes (RP2, STK19P, CYP21A1P y TNXA) denominados así por su homología20.
El gen (CYP21A2) y el pseudogen (CYP21PA1) son idénticos en el 98% de su secuencia de nucleótidos. El pseudogen posee varias mutaciones que inactivan por completo su producto génico. La mayoría de las mutaciones que causan el 21OHD surgen a partir de dos tipos de recombinación entre el gen y el pseudogen:
- •
Conversión de genes (75%): transferencia de mutaciones del pseudogen al gen durante la mitosis.
- •
Recombinación meiótica (25%): se elimina una pequeña secuencia de nucleótidos correspondiente a un segmento de 30kb de gen que abarca el extremo 3’ del pseudogen CYP21P, la totalidad del gen del complemento C4B adyacente y el extremo 5’ de CYP21A2, produciendo un pseudogen quimérico no funcional.
El 1-5% restante corresponde a mutaciones de novo u otras puntuales20.
Correlación genotipo-fenotipoLas mutaciones de CYP21A2 pueden clasificarse en tres categorías según el nivel de actividad enzimática predicha a partir de la mutagénesis in vitro y los estudios de expresión, definiendo el fenotipo para cada tipo de mutación (tabla 3)17,21. Sin embargo la correlación genotipo-fenotipo no es estricta, New et al. analizaron el DNA de 1.507 familias con al menos un miembro con HAC, encontrando que los fenotipos de pérdidas salinas y HACNC tienen buena correlación genotípica, no así para la variedad virilizante simple de la cual reportan gran variabilidad. Concluyen que la correlación directa solo se da en menos del 50%22. Los resultados anteriores contrastan con lo encontrado por Bidet et al. quienes caracterizaron molecularmente a 161 pacientes con HACNC encontrando gran variabilidad genotípica23–25.
Correlación genotipo, actividad enzimática y fenotipo
| N.°. | Mutación | Actividad enzimática | Forma clínica |
|---|---|---|---|
| 1 | Nonsense o deleciones | Abolición total | PS |
| 2 | Missense | 1-2% actividad normal | VS |
| 3 | Val281Leu Pro30Leu | 20-50% | HACNC |
HACNC: hiperplasia adrenal congénita no clásica; PS: pérdidas salinas; VS: virilizante simple.
HAC virilizante simple: es evidente desde el momento del nacimiento, ya que se presenta alteración en la morfogénesis genital a causa de un hiperandrogenismo en el feto desde la séptima semana de vida intrauterina10. Los recién nacidos de género femenino presentan virilización de los genitales externos en diferentes grados y se clasifican según la escala de Prader, la cual se numera de grados I-V: I) clitoromegalia sin fusión labial; II) fusión posterior de labios; III) mayor clitoromegalia con orificio urogenital perineal y fusión labial casi completa; IV) clítoris fálico, orificio urogenital en base del clítoris y fusión labial completa y V) clítoris con forma peneal, orificio uretral en punta del falo y labios totalmente unificados con aspecto de escroto, puede presentar uretra oculta dentro del falo y causar confusión con un paciente masculino con criptorquidia bilateral e hipospadias. Sin embargo, los derivados mullerianos (útero, ovarios y trompas de Falopio) no presentan alteraciones. La presencia de genitales ambiguos no confirma el diagnóstico por lo cual se debe tener en cuenta otras enfermedades que puedan cursar con esta presentación26–29.
En contraste, en los hombres es más difícil identificar la enfermedad al nacimiento ya que los signos clínicos son muy sutiles, presentando hiperpigmentación melánica no racial del escroto por aumento de la hormona melanocítico-estimulante y pene más grande de lo normal, signos que en conjunto se denominan macrogenitosomía y que después se asocian con pubertad precoz. Las mujeres manifestarán además de pubertad precoz, hirsutismo, oligomenorrea o amenorrea, ovarios poliquísticos y acné. En ambos sexos se presentará crecimiento acelerado y baja estatura final por cierre prematuro de las epífisis óseas30.
HAC con pérdidas salinas: presenta manifestaciones clínicas desde el nacimiento. Al haber deficiencia total de aldosterona habrá un trastorno hidroelectrolítico por incapacidad de retener sodio, agua y excretar potasio, resultando en una crisis adrenal, es decir, una deficiencia de aldosterona sintomática, definida como un episodio de hiperpotasemia, hiponatremia y acidosis metabólica, manifestada con inapetencia, letargia, vómito, diarrea, poliuria, deshidratación, pérdida de peso, hipotensión y choque hipovolémico. Muchas veces es esta la forma clínica con la que comienzan y de no ser tratado puede llevar a la muerte en pocas h31.
Además, las concentraciones elevadas de 17OHP actúan como antagonista de mineralocorticoides, exacerbando así los efectos del déficit de aldosterona por sus efectos natriuréticos los cuales se evidencian de la primera a la cuarta semana de vida extrauterina1,21.
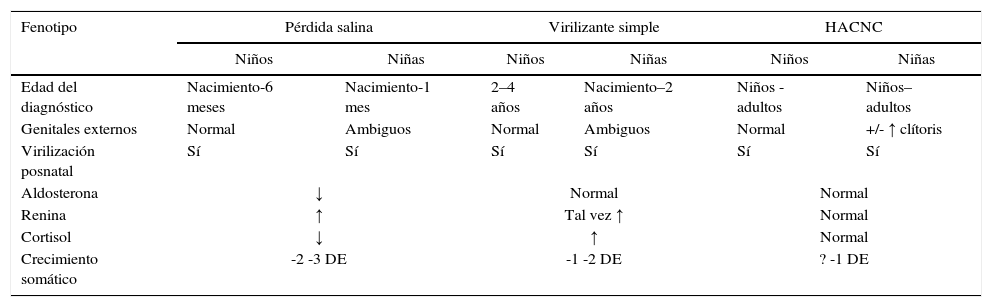
Hiperplasia adrenal congénita no clásica: estos pacientes presentan concentraciones normales de cortisol y exceso de andrógenos pero sin deficiencia de aldosterona10. Las manifestaciones clínicas aparecen en cualquier momento de la vida y corresponden a grados variables de pubarquia precoz (presencia de vello púbico, vello axilar y olor apocrino antes de los 8 años en niñas y 9 años en niños), hirsutismo (crecimiento excesivo de vello terminal en la mujer siguiendo un patrón de distribución masculino)32, acné quístico, hipertrofia del clítoris, aceleración del crecimiento y de la maduración ósea, irregularidades menstruales y ovario poliquístico. En hombres calvicie, oligospermia e infertilidad5,7,30. La tabla 4 resume la presentación clínica de los pacientes con HAC.
Presentación clínica
| Fenotipo | Pérdida salina | Virilizante simple | HACNC | |||
|---|---|---|---|---|---|---|
| Niños | Niñas | Niños | Niñas | Niños | Niñas | |
| Edad del diagnóstico | Nacimiento-6 meses | Nacimiento-1 mes | 2–4 años | Nacimiento–2 años | Niños - adultos | Niños–adultos |
| Genitales externos | Normal | Ambiguos | Normal | Ambiguos | Normal | +/- ↑ clítoris |
| Virilización posnatal | Sí | Sí | Sí | Sí | Sí | Sí |
| Aldosterona | ↓ | Normal | Normal | |||
| Renina | ↑ | Tal vez ↑ | Normal | |||
| Cortisol | ↓ | ↑ | Normal | |||
| Crecimiento somático | -2 -3 DE | -1 -2 DE | ? -1 DE | |||
DE: desviaciones estándar; HACNC: hiperplasia adrenal congénita no clásica.
Tomado y modificado de: White PC, Speiser FW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine Reviews. 2000;21:245-91.
El abordaje inicial después del examen físico exhaustivo es con un cariotipo (para determinar el sexo genético del paciente), niveles de 17OHP, electrólitos, actividad de renina plasmática, androstenediona y progesterona5. Se recomienda realizar un ultrasonido abdomino-pélvico para identificar el útero y evaluar el tamaño adrenal10,11.
Diagnóstico hormonal: en el déficit de 21OHD la concentración plasmática de 17OHP está aumentada. Por esta razón, hay que tener en cuenta que puede haber falsos positivos ya que este precursor suele estar aumentado al nacer y disminuye en forma progresiva en neonatos no afectados, por esto la sensibilidad diagnóstica es pobre en los primeros dos días después del nacimiento11. Se recomienda que las pruebas sean tomadas antes de las 8:00 am, teniendo en cuenta el ciclo circadiano de la síntesis y secreción de cortisol. En los pacientes con elevación de 17OHP es necesario solicitar electrólitos, glucemia, gases venosos y otras hormonas como progesterona, androstenediona y en algunos casos testosterona y 21-desoxicortisol, cuyos valores podrían estar alterados8,10,19.
Forma clásica: en neonatos sanos los niveles de 17OHP son inferiores a 20ng/mL; la prueba será positiva si se obtienen niveles séricos mayores a este en las primeras 48 h de vida. Si están ligeramente elevados se debe repetir la prueba, pero si los valores están muy elevados o hay inestabilidad clínica, se requiere una evaluación urgente con toma de electrólitos, 17OHP y seguimiento clínico8,19,33.
Pérdida salina: se evidencian cifras elevadas de renina y disminución de la relación aldosterona/renina, la concentración de 17OHP suele ser muy alta, por lo general hasta de 1.000ng/mL, los niveles séricos de sodio son menores de 110 mEq/lt y de potasio mayores de 10 mEq/lt, siendo un reflejo del déficit de aldosterona. Pueden presentar también acidosis metabólica e hipoglucemia10,33.
Virilizante simple: la sospecha diagnóstica parte de las manifestaciones clínicas propias de la virilización, lo que se correlaciona con la concentración de 17OHP, que estará mayor de 100ng/mL. Además, puede existir cierto grado de déficit en la síntesis de aldosterona y por esto elevación de las concentraciones plasmáticas de renina y de potasio.
Forma no clásica: la concentración plasmática de 17OHP puede estar normal o con ligera elevación, por ello el diagnóstico se realiza con la prueba de estimulación con ACTH, en la cual se cuantifica la concentración basal de 17OHP, después se administran 250μg/m2 de ACTH y pasados 30-60 min se cuantifica de nuevo la 17OHP. Algunos autores la consideran positiva cuando los valores superan los 5ng/mL (15 nmol/l) y 15ng/l (45 nmol/l) para las mediciones pre- y postestímulo respectivamente. Sin embargo tomando como punto de corte 14ng/mL (42 nmol/l) se obtiene una sensibilidad del 90,9% y una especificidad del 100%, por tanto, si se tomara como punto de corte se escaparían del diagnóstico el 10% de los pacientes con HACNC (falsos negativos), por lo cual recomiendan tomar como punto de corte 10ng/mL (30 nmol/l). Valores entre 10-15ng/mL son sugestivos pero no concluyentes, por tanto, requieren repetir la prueba o realizar estudios genéticos33,34.
Diagnóstico prenatal: está dirigido a familias en las que exista un primer miembro diagnosticado o en parejas en las que se sepa el estado de portadores. El diagnóstico del feto con riesgo se puede hacer al final del primer trimestre del embarazo por medio del análisis de ADN obtenido por biopsia de las vellosidades coriónicas o en el segundo trimestre, alrededor de la 15 a 18 semanas de gestación cuantificando la concentración de 17OHP en líquido amniótico21,35.
Los nuevos procedimientos de diagnóstico prenatal no invasivos se realizan mediante la extracción de muestras de ADN fetal libre en el plasma materno mediante la secuenciación del gen CYP21A2 para identificar fetos femeninos que pueden ser afectados entre las semanas 6-7 de gestación. En forma ideal se espera la aplicabilidad de los métodos no invasivos previos a la sexta semana de gestación para iniciar manejo precoz. La aplicación de métodos diagnósticos no invasivos para la intervención temprana y consiguiente prevención de ambigüedad sexual, previene la intervención quirúrgica en recién nacidos afectados y, a futuro, simboliza una conservación en la función sexual del individuo36.
Tamizaje neonatalDesde 1981 el tamizaje neonatal es recomendado por la Sociedad Europea de Endocrinología Pediátrica, se realiza mediante la evaluación de 17OHP en sangre total. El tamizaje reduce la mortalidad en neonatos de sexo masculino que en ausencia de tratamiento es del 75%, además previene errores en asignación del sexo en mujeres y facilitar el diagnóstico en hombres8,11,14,18,37,38.
La concentración de 17OHP sugestiva de 21OHD para neonatos de 3-5 días de vida son: 133ng/mL (400 nmol/l) en recién nacidos menores de 35 semanas de gestación y 150 nmol/l en recién nacidos de 35 a 36 semanas. Se ha visto que la tasa de falsos positivos al cuantificar la 17OHP con el método inmunológico es alta, probablemente por reacción cruzada con otros esteroides o porque las muestras se toman muy pronto39. Los neonatos prematuros también pueden tener concentraciones más altas de ACTH como resultado de estrés y pobre función adrenal, lo cual es otro factor importante. Por ello, la recomendación es que todos los recién nacidos pretérmino o con bajo peso al nacer deben ser tamizados entre las 48 y 72 h del nacimiento y luego a los 28 días de vida. Los neonatos que reciben tratamiento prenatal con glucocorticoides para inducir la maduración pulmonar pueden tener bajas concentraciones de 17OHP durante la primera semana de vida, siendo causa de falsos negativos, por lo que se debe cuantificar de nuevo a los 14 días de vida. Para disminuir la tasa de falsos positivos, es necesario relacionar los niveles plasmáticos de 17OHP con la edad gestacional y el peso al nacer, además de realizar procedimientos de extracción con éter para minimizar la interferencia de los metabolitos en el resultado de las pruebas38,40–42.
Sin embargo el rendimiento del tamizaje inicial no es ideal. El valor predictivo positivo del tamizaje neonatal es <1% lo que significa que por cada 100 muestras que son positivas, solo una podría representar un verdadero caso de HAC. Por eso, se han llevado a cabo varias pruebas de segunda línea para aquellos pacientes con tamizaje positivo, descartando que sean falsos positivos. La prueba de segunda línea recomendada es la cromatografía líquida–espectrometría de masa en tándem (HPLC-TMS) en la que se incluyen otros metabolitos como el cortisol y la androstenediona, y se sigue midiendo la 17OHP en muestras recogidas en papel filtro. En algunos programas la genotipificación es usada como pruebas de segundo nivel43–45.
Tratamiento médicoSe basa en la terapia de remplazo hormonal con glucocorticoides en pacientes con HACC y la forma sintomática de la HACNC; en aquellos con pérdidas salinas se deben añadir mineralocorticoides, suprimiendo así por retroalimentación negativa la secreción de CRH y ACTH y por ende, la estimulación adrenal46.
Tratamiento prenatal: en madres gestantes de un feto con riesgo de tener HAC se debe iniciar el tratamiento con el fin de reducir la virilización femenina y por tanto la necesidad de cirugía reconstructiva. Debido a que los genitales externos son virilizables desde la octava semana de gestación y el diagnóstico por biopsia de vellosidades coriónicas solo se puede realizar desde la duodécima semana, el tratamiento debe iniciarse en forma empírica tan pronto como se confirme el embarazo. Una vez se realice la biopsia y se confirme o descarte la enfermedad, se decide si continuar o no con el tratamiento. Cerca de 85% de los fetos de sexo femenino que reciben terapia nacen con genitales normales o con leve virilización según la escala de Prader3,47.
El medicamento de elección es la dexametasona ya que esta escapa de la inactivación por la 11β-hidroxiesteroide deshidrogenasa placentaria tipo II, permitiendo su paso transplacentario y por tanto, concentraciones terapéuticas efectivas en el feto. La dosis usada es de 20μg/k/día dividido en tres tomas, ya que una sola dosis al día no es efectiva para mantener niveles plasmáticos eficaces durante el día48–50.
Tratamiento de pacientes en crecimiento: el objetivo es disminuir la sintomatología, normalizar la velocidad de crecimiento y la tasa de maduración esquelética, alcanzar la pubertad en el momento adecuado, y en adolescentes y mujeres adultas regularizar los ciclos menstruales y alcanzar tasas de fertilidad óptimas5.
En los pacientes en crecimiento el corticoide de elección es la hidrocortisona ya que es el glucocorticoide con menor vida media, lo cual disminuye el riesgo de las reacciones adversas, sobre todo la talla baja. Los casos con HACC requieren dosis entre 15-20mg/m2/día de hidrocortisona en tabletas, repartidas en tres dosis. Aquellos con la forma virilizante simple requieren entre 8-10mg/m2/día33. Hay divergencia de opiniones en administrar tres dosis iguales durante el día, o que la dosis de la mañana sea mayor que las otras dos, tratando de simular la secreción fisiológica, pero la evidencia señala que existe poca diferencia entre las dos. Sin embargo, se ha desarrollado una presentación de hidrocortisona de liberación modificada (MR-HC)-Chronocort. Estudios clínicos en fase 2 sugieren que el uso de esta forma de hidrocortisona imita mejor los pulsos de cortisol fisiológicos y disminuye la concentración matutina de 17OHP. También se ha implementado la infusión de hidrocortisona por medio de una bomba programable e infusiones subcutáneas continuas de hidrocortisona51.
Cuando el paciente entra en la pubertad hay hiperandrogenismo fisiológico, por eso se debe evaluar la terapia en cada etapa de la vida ya que en esta podría requerir dosis mayores46, teniendo como objetivo encontrar la dosis más baja del glucocorticoide con la que se suprima el eje, para mantener un equilibrio entre el tratamiento y las complicaciones después de la exposición prolongada a glucocorticoides. Por lo regular esta dosis es menor a 17mg/m2/día18. Hay que tener en cuenta que las dosis requeridas varían entre individuos, lo cual representa una base genética relacionada con polimorfismos en la enzima 11β-hidroxiesteroide deshidrogenasa que podrían modular la respuesta a los glucocorticoides52,53.
En pacientes adultos con HAC pueden formularse corticoides de acción corta, media y larga, recomendándose el uso de una cantidad diaria total de dexametasona (0,5mg/día), prednisolona (4,75mg/día) o hidrocortisona (13,75mg/m2/día) repartida en tres dosis18.
Además del glucocorticoide, los pacientes que padecen la forma clásica con pérdidas salinas deben recibir fludrocortisona en dosis de 0,05-0,3mg/día en dos o tres tomas, más un suplemento de sodio con las comidas (2g/día). La dosis del mineralocorticoide debe ser evaluada a medida que crece el paciente, pues la sensibilidad cambia con la edad y según el glucocorticoide usado, ya que todos tienen diferente actividad mineralocorticoide. El uso de mineralocorticoides permite disminuir los requerimientos del glucocorticoide y por tanto sus reacciones adversas54.
Los exámenes de laboratorio y el examen clínico son indispensables para seguir el tratamiento. Por eso deben cuantificarse cada tres meses en la infancia y entre 4 y 12 meses después de ella: los electrólitos séricos, 17OHP, androstenediona y la actividad de renina plasmática9.
Manejo de la crisis adrenal: la crisis adrenal es una urgencia médica y se ha clasificado según su severidad, en la cual se basa la conducta terapéutica como ilustra la tabla 5.
Clasificación y manejo de la crisis adrenal
| Severidad | Manifestaciones cínicas | Tratamiento |
|---|---|---|
| Leve | Deterioro general, pero tolera fluidos | Las dosis del glucocorticoide y fludrocortisona deben duplicarse |
| Moderada | Vómito y signos de deshidratación | Hidrocortisona intramuscular |
| Severa | Alteración del estado de conciencia, hipotensión | • Bolo de solución salina 0,9% 20ml/k. • Bolo hidrocortisona 2ml/k. • Infusión de hidrocortisona |
Dosis de estrés: los dos sistemas de respuesta al estrés (producción cortical de cortisol y medular de catecolaminas) tienen una relación ontogénica, anatómica y fisiológica íntima55. Los pacientes con un déficit severo de 21OH o aquellos bajo tratamiento prolongado con glucocorticoides son incapaces de producir cortisol en respuesta al estrés físico, por eso necesitan dosis extra en estas situaciones. La conducta es duplicar o triplicar la dosis del glucocorticoide oral con la que el paciente viene siendo manejado. Además, los pacientes con HAC suelen tener algún grado de disfunción medular por lo cual están en riesgo de desarrollar hipoglucemia durante el periodo de estrés, por lo que se debe asegurar un aporte calórico adecuado y un control periódico de la glucemia. Considerando que los pacientes con HACNC presentan concentraciones subóptimas de cortisol tras el estímulo con ACTH y que no tienen presentación clínica clara de insuficiencia adrenal, debe usarse dosis de estrés cuando estén sometidos a trauma extremo o cirugía mayor34.
Tratamiento quirúrgico: el manejo quirúrgico es decir la genitoplastia, se recomienda entre los 2-6 meses de edad para las niñas con grado III o más en la escala de Prader, ya que los tejidos son más adaptables y moldeables, además minimizará el trauma psicológico para la paciente y sus padres18. El procedimiento consiste en la eliminación de tejido eréctil fálico (clitoroplastia) y en realizar una abertura vaginal de separación funcional de la uretra y la vagina (vaginoplastia). La complicación más importante tras el procedimiento es la estenosis vaginal que puede llegar a requerir dilatación o reintervención quirúrgica.
Debido a la sobreproducción de andrógenos adrenales, la adrenalectomía es una buena alternativa para suprimir la síntesis de estas hormonas. El procedimiento bilateral lo recomiendan algunos investigadores quienes opinan que tratar la enfermedad de Addison es menos problemático que el manejo de la HAC, ya que las dosis del glucocorticoide que se usan son menores. Los pacientes con características combinadas de hiperandrogenismo (a pesar del tratamiento médico adecuado) y efectos deletéreos por el manejo con glucocorticoides, pueden beneficiarse del procedimiento. Las potenciales complicaciones, además del riesgo quirúrgico, son baja o nula respuesta al estrés debido a la deficiencia de epinefrina y el riesgo de desarrollar el síndrome de Nelson (tumor hipofisario productor de ACTH). Por lo anterior la cirugía debe considerarse como último recurso en pacientes que han fallado con la terapia médica, en especial en las mujeres con pérdidas salinas y características de hiperandrogenismo severo e infertilidad. Es poca la información que aporta la literatura, pero algunas series de casos sugieren que es un procedimiento seguro, con buena respuesta que representa beneficios en la calidad de vida y el estado de salud de los pacientes56,57.
Nuevas terapiasLa Sociedad de Endocrinología recomienda que en todo niño con una talla predicha final de -2,25 DE o mayor hay que considerar el tratamiento experimental en estudios controlados bien diseñados11. Las nuevas terapias están encaminadas a mejorar el crecimiento y la evolución a largo plazo. Existen medicamentos que bloquean el receptor de andrógenos como el acetato de ciproterona, flutamida y otros medicamentos derivados de ella como la nilutamida y la bicalutamida, usados principalmente en el tratamiento del cáncer de próstata. Estos tienen mejores propiedades farmacocinéticas que la flutamida, lo que permite que su administración sea una sola vez al día, disminuyendo el riesgo de hepatotoxicidad, pero no han sido estudiados en pacientes con HAC, por lo cual su eficacia clínica en esta enfermedad es incierta. Algunos autores sugieren su uso en mujeres con hirsutismo o calvicie androgénica resistente a las otras terapias. El acetato de ciproterona tiene un perfil de mejoría más alto para manejar el hirsutismo comparado con la hidrocortisona56.
Se han llevado a cabo estudios preclínicos y en fase I con acetato de abiraterona, un potente antiandrógeno que inhibe en forma irreversible con gran selectividad y afinidad las enzimas 17 alfa hidroxilasa, 17,20 liasa necesarias para la síntesis de andrógenos. Este es un medicamento prometedor para los pacientes con HAC, sin embargo hacen falta más estudios para determinar su seguridad y eficacia clínica. También se ha implementado el uso de antiandrógenos (flutamida) e inhibidores de la conversión de andrógenos a estrógenos (testolactona), ya que los pacientes con HAC cursan con un hiperestrogenismo que afecta el crecimiento óseo. Los resultados preliminares de un experimento clínico mostraron que la testolactona y flutamida reducen las alteraciones de la edad ósea, el peso y la velocidad de crecimiento en comparación con el tratamiento habitual de hidrocortisona y fludrocortisona. Algunos autores consideran que la instauración de un régimen de cuatro medicamentos: dosis bajas de glucocorticoide, mineralocorticoide, testolactona y flutamida reducen la tasa de crecimiento en la edad ósea y lentifica la ganancia de peso18.
Buscando mejorar la talla final, se han venido usando análogos de la hormona liberadora de gonadotrofinas (GnRHa) junto con GH que impiden la progresión de la pérdida de estatura, mejorando la velocidad de crecimiento y la talla final después de dos años de tratamiento. Sin embargo, estos medicamentos no se usan de forma sistemática para el tratamiento de la HAC58.
Terapia génica: se han realizado estudios en la línea de murinos recombinantes H-2aw18 introduciendo fragmentos de ADN recombinante con el gen murino para la 21OH, mostrando que esta línea de roedores es válida como modelo de estudio. Los adenovirus son un buen vector para la terapia génica en HAC ya que tienen un tropismo selectivo por la glándula adrenal animal. Se han realizado estudios con inyecciones intraadrenales usando adenovirus como vector del gen que codifica para la 21OH, y con el seguimiento concluyen que este método mejora la función adrenocortical en roedores con 21OHD, sin embargo no se han realizado estudios en humanos56.
Complicaciones y situaciones especialesLos pacientes con HAC tienen una serie de complicaciones relacionadas con la enfermedad o su tratamiento, como sucede con la talla final, que puede afectarse por el déficit enzimático o por el tratamiento con glucocorticoides. La mayoría de los pacientes no alcanzan la talla predictiva final ni el objetivo de altura esperado con respecto a la talla de sus padres59. El paciente con HAC, como todos los que requieren uso crónico de corticoides, tiene aumentado el riesgo de síndrome metabólico, osteopenia y osteoporosis. Por esta razón, debe considerarse el uso profiláctico vitamina D, suplementos de calcio, nutrición apropiada y actividad física específica60–62.
En el 20% de los pacientes con HAC existe un remanente adrenal en los testículos, que al someterse a concentraciones elevadas de ACTH se hipertrofian generando un tumor testicular de restos adrenales (TARTs) que causa obstrucción del túbulo seminal y por consiguiente infertilidad. Si esto sucede se debe intensificar la terapia con corticoides (dexametasona 0,75-2mg/día) para disminuir el tamaño, pero si el manejo médico fracasa se recomienda la extirpación quirúrgica18.
La tasa de embarazos en mujeres con HAC es baja, lo que puede ser secundario al retraso en el desarrollo psicosexual, diferencias en la orientación psicosocial, poca actividad sexual, sobreproducción adrenal de andrógenos y pobre resultado quirúrgico (introito vaginal inadecuado), sin lograr corregir todos los defectos sexuales. Además estas pacientes cursan con grados variables de dispareunia y dificultad para lograr el orgasmo. Cuando cursan con embarazo deben continuar la terapia con corticoides (hidrocortisona, prednisona o prednisolona); y teniendo en cuenta que el trabajo de parto es un evento que demanda una respuesta fisiológica al estrés, se debe suplir la dosis de estrés durante este63.
El hecho de estar expuesto a concentraciones elevadas de andrógenos durante el período pre- y posnatal puede afectar el desarrollo cerebral en niñas, que en el futuro suelen tener un comportamiento sexual atípico, poco interés en gustos propios del género y la edad, por ejemplo jugar con muñecas. En cuanto a la orientación sexual, las tasas de homosexualidad y bisexualidad son menos consistentes y varían entre el 5-37%. Sin embargo, hay que tener en cuenta que los factores sociales, familiares, educativos y emocionales también influyen en el desarrollo del género.
ConclusiónLa HAC es una entidad de gran variabilidad clínica y paraclínica que exige en el médico (especialista o no) un amplio conocimiento en cuanto a la forma de presentación, diagnóstico y manejo en situaciones especiales (crisis adrenal, dosis de estrés y embarazo), además de estar capacitado para realizar el seguimiento regular, las intervenciones tempranas en el estilo de vida, la valoración de la salud ósea, de la ecografía testicular y el manejo psicológico. Es fundamental recordar que el diagnóstico y tratamiento oportuno a veces no son suficientes, pues se debe enfocar al paciente como un todo, desde una perspectiva holística e individualizada con el objetivo no solo de prescribir medicamentos, solicitar exámenes, aliviar síntomas o dar un pronóstico favorable, sino de entender al individuo como un ser pensante y sensible. Algunas veces una conversación es la mejor catarsis para cualquier enfermedad. De acuerdo a la revisión realizada, no se conocen datos a nivel local con respecto a la deficiencia de 21OH. La HAC es una condición de baja frecuencia pero de gran impacto en la morbimortalidad de los neonatos, conocer su comportamiento en la población latinoamericana ayudará a entender su historia natural y orientará al desarrollo de nuevas terapias.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.