



Equine influenza virus is a leading cause of respiratory disease in horses worldwide. Disease prevention is by vaccination with inactivated whole virus vaccines. Most current influenza vaccines are generated in embryonated hens’ eggs. Virions are harvested from allantoic fluid and chemically inactivated. Although this system has served well over the years, the use of eggs as the substrate for vaccine production has several well-recognized disadvantages (cost, egg supply, waste disposal and yield in eggs). The aim of this study was to evaluate a baculovirus system as a potential method for producing recombinant equine influenza hemagglutinin to be used as a vaccine. The hemagglutinin ectodomain (HA1 subunit) was cloned and expressed using a baculovirus expression vector. The expression was determined by SDS-PAGE and immunoblotting. A high yield, 20μg/ml of viral protein, was obtained from recombinant baculovirus-infected cells. The immune response in BALB/c mice was examined following rHA1 inoculation. Preliminary results show that recombinant hemagglutinin expressed from baculovirus elicits a strong antibody response in mice; therefore it could be used as an antigen for subunit vaccines and diagnostic tests.
El virus de la influenza equina es una de las principales causas de enfermedad respiratoria en caballos de todo el mundo. La prevención de la enfermedad es a través de la vacunación con vacunas a virus inactivado. La mayoría de las vacunas se producen en huevos embrionados, de los cuales los viriones son cosechados del líquido alantoideo e inactivados químicamente. Aunque este sistema ha servido bien durante años, el uso de huevos como sustrato para la producción de vacuna presenta varias desventajas bien reconocidas (costo, provisión de huevos, manejo de los residuos, rinde por huevo). El objetivo del presente trabajo fue evaluar preliminarmente un sistema de expresión en baculovirus como método de producción de hemoaglutinina recombinante (rHA) para ser utilizada como vacuna para la prevención de la influenza equina. Para ello el ectodominio de la hemaglutinina (la subunidad HA1) del virus de la influenza equina se expresó en células de insecto infectadas con un baculovirus recombinante. La expresión fue demostrada por SDS-PAGE e inmunoblotting. El método empleado fue capaz de producir gran cantidad de rHA1. En este estudio se obtuvieron 20μg/ml (200μg de HA1 purificada de 2,5x107 células infectadas). La respuesta inmune fue evaluada mediante la inmunización de ratones BALB/c. Los resultados preliminares demostraron que la proteína recombinante expresada en baculovirus genera una fuerte respuesta inmune en ratones, por lo tanto podría ser utilizada como antígeno para la producción de una vacuna a subunidades y en pruebas diagnósticas.
Equine Influenza is a disease that affects horses, donkeys and mules, which is currently considered one of the main causes of equine respiratory diseases around the world. Its causative agent is a virus belonging to the Orthomyxoviridae family. Conventional equine influenza vaccines contain inactivated whole viruses either as an aqueous suspension or combined with adjuvants such as aluminum hydroxide or aluminum phosphate. For vaccine production, the influenza virus is usually grown in the amniotic cavity of fertile hens’ eggs or in a cell culture maintained in medium containing trypsin. Virions are harvested from egg allantoic fluid or culture medium and are inactivated or treated with detergent. A basic disadvantage these systems present is their variability and the heterogeneity of their cellular constituents9,13. Therefore, other methods for equine influenza vaccine production were developed. These include a canarypox-vectored vaccine (ProteqFlu, Merial Inc., USA), a subunit vaccine containing purified hemagglutinin and neuraminidase proteins adjuvated with ISCOMatrix (Equilis Prequenza), an attenuated live vaccine (Flu AvertTM I.N., Intervet) and many inactivated influenza vaccines, with the latter being the most widely used commercially. Vaccination against equine influenza has been recently reviewed12. During 2008, Protein Sciences Corporation reported the development of a human influenza vaccine, named FluBlok®, using the insect cell/baculovirus technology. This vaccine was shown to be safe and efficacious in clinical trials3,4,15. Insect cell systems have been widely used to produce recombinant proteins. The high yield obtained by baculovirusinfectedinsect cells makes them attractive tools for pharmaceutical protein production6.
Hemagglutinin is the dominant glycoprotein on the surface of the influenza virus and a recognized key antigen in the host response to influenza virus in both natural infection and vaccination17. The protein, encoded by segment 4 of the viral genome, is initially translated as a preprotein (HA0) that is then split into two subunits (HA1 and HA2), which subsequently assemble to form a trimeric structure (HA1-HA2). In the spatial conformation that the mature hemagglutinin takes, only the HA1 subunit becomes exposed and it is, therefore, where most of its antigenic determinants are found16.
The present study was aimed at evaluating a baculovirus expression system as a potential method for producing recombinant equine influenza hemagglutinin.
Materials and methodsVirus and cellsAn Argentine equine influenza virus strain previously isolated in our laboratory, sharing high homology to strain A/ equine/Argentina/1999 (H3N8) (more than 99%) and closely related to Floride clade 1 strains, was propagated in the allantoic cavity of 11 day-old embryonated hen's eggs.
Spodoptera frugiperda (Sf9 and Sf21) cells (Gibco, Grand Island, NY, USA), used for the transfection and propagation of recombinant viruses, were cultured at 27°C in TC-100 media (US Biological, Swampscott, MA, USA) supplemented with 10% Fetal Bovine Serum (FBS) (PAA Laboratories GmbH, Pasching, Austria).
For the expression of recombinant HA1 subunit, Sf9, Sf21 cells and Trichoplusia ni High Five cells (Invitrogen, Carlsbad, CA, USA) were used. Three different culture media were tested: Grace's (Gibco), TC-100 and SF900II (Gibco).
Amplification by RT-PCRTotal RNA was extracted using Trizol reagent (Invitrogen). The reverse transcription was carried out using 1 μg of total RNA, the HA1R primer: 5° CACTCGAGTTGCTTTTCTGGTACATTCCTC 3° and Moloney Murine Leukemia Virus Reverse Transcriptase (Promega, Madison,WI, USA) under conditions specified by the supplier. The PCR reaction was performed by adding 5μl from the cDNA to a reaction mixture containing a final concentration of 200μM of each one of the dNTPs, 1.5mM of MgCl2, 0.25U of Taq DNA polymerase (Fermentas Inc, Glen Burnie, MD, USA) and 10 pmol of each one of primers: HA1R and HA1F: 5° TACCATGGTCTACAGTCAAAACCCAACCAG 3°. The amplification was performed on a Mastercycler gradient (Eppendorf, Hamburg, Germany). After denaturation at 94°C for 2m, the reactions were cycled 30 times at 94°C for 30 s, 63°C for 30 s and 72°C 1m. This was followed by a final elongation step at 72°C for 10m. The PCR product was run on 1.5% agarose gel dyed with ethidium bromide.
Construction of a baculovirus transfer vectorThe PCR-amplified HA1 segment was ligated to a pCR2.1TOPO vector (Invitrogen) and transformed into One Shot TOP10 Competent Cells (Invitrogen). Recombinant clones containing the insert were examined to ensure proper orientation by endonuclease restriction with XhoI (Promega) and DNA sequence analysis (Biotechnology Resource Center - Cornell University - Ithaca - New York, USA). For sub cloning, the HA1 coding sequence was digested with NcoI and BamHI (Promega). The resulting plasmid named pFastBac-HA was used to generate a recombinant baculovirus.
Generation of a recombinant baculovirusIn order to induce transposition between the recombinant transfer vector and Autographa californica nuclear polyhedrosis virus bacmid (bAcNPV), the pFastBac-HA vector was transformed into Eschericia coli DH10Bac (Invitrogen). After incubation, white recombinant and blue non-recombinant colonies could be clearly differentiated. To confirm the transposition of the HA1 segment into the bAcNPV, 12 white colonies and one blue colony were analyzed for Colony- PCR with M13 universal primers according to the protocol included in the Bac to Bac kit manual (Invitrogen).
The recombinant bacmid DNA extracted from previously tested white colonies was used to transfect Sf21 cells by using Insect GeneJuice® Transfection Reagent (Novagen, Madison, WI, USA). The transfected cells were cultured at 27°C in TC-100 media supplemented with 10% fetal bovine serum. A primary virus stock was collected at 96h posttransfection and used to perform another two rounds of infections. At 72h of the second infection, the culture medium supernatant was stored at –70°C to be used as viral stock in future protein expression assays.
Expression of recombinant proteinsHigh-titre recombinant baculovirus stock was used to infect three insect cell lines (Sf9, Sf21 and High Five). Cell growth profile was carried out at 27°C in 25cm2 cell culture flasks (Greiner Bio-one, Frickenhausen, Germany) with three different media: TC-100 and Grace's supplemented with 10% FBS and SF-900 II with and without FBS. Cells were infected during the mid-exponential growth phase, using a multiplicity of infection (MOI) of 0.25. A preliminary comparison of protein yields obtained with the three media was conducted by sampling 100μl of culture supernatants at different post-infection times. Supernatants from each cell line were collected daily during 4 days and recombinant HA1 production was analyzed by Polyacrylamide Gel Electrophoresis (SDS-PAGE). The proteins were separated by standard nonreduced SDS-PAGE on a 5% stacking gel and 12.5% polyacrylamide gel for 3.5 h at 60 V. The gels were stained with 0.1% Coomassie Brilliant Blue R250 (Sigma).
Purification of the HA1 proteinA high-yield, homogenous preparation of HA1 subunit was obtained by using the nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen, Hilden, Germany), according to standard procedures. In brief, after finding the optimal expression conditions (see Results) a new culture at larger volume (25ml of culture medium with 106 cells/ml) was made in 175cm2 cell culture flasks (CellStar – Greiner). Three days post-infection, cells were harvested and washed three times with sterile PBS. The whole pellet of cells was disrupted by sonication with Binding Buffer (50mM NaH2PO4, 500mM NaCl, pH 8.0).
The cell lysate was pelleted at 5000g per 10m and the total protein solution present in the supernatant (20ml) was loaded into a Ni-NTA column pre-equilibrated with Binding Buffer. The bound protein was washed with buffer 50mM NaH2PO4, 500mM NaCl, 20mM Imidazole, pH 8.0 until the absorbance at an OD of 280nm returned to baseline. Once the baseline was stable, elution of the bound recombinant protein was carried out using 20ml of 50mM NaH2PO4, 500mM NaCl, 250mM Imidazole (Merck), pH 8.0. The eluted recombinant protein was concentrated by precipitation with absolute etanol and resuspended in 10ml of sterile PBS.
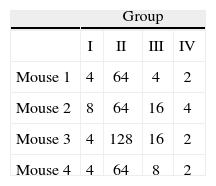
Immunization of mice with HA1 proteinSixteen 6 week- old female BALB/c mice were randomized into four groups. All animal procedures were performed conforming to approved protocols and in accordance with recommendations for the proper use and care of laboratory animals. Group I was intranasally (i.n.) inoculated with 1μg of purified protein (quantified by the Bradford assay). Group II was inoculated intramuscularly (i.m.) with a mixture of 25μl of HA1 protein solution (500ng of purified protein) and 25μl of Specol adjuvant (ID-DLO, Lelystad, The Netherlands). Group III was inoculated i.m. with 50μl of HA1 protein solution (1μg of purified protein). Group IV was used as non-immunized control (table 1). Two booster immunizations were performed 15 and 30 days post-priming in identical conditions, but without adjuvant in the Group II. Serum samples of each group were collected and pooled before each inoculation and analyzed by Haemaglutination inhibition test (HI). Fifteenth days after the last immunization all mice were sacrificed and the level of anti-HA1 antibodies was estimated by Western blot (WB) and HI from the pooled and individual samples respectively. The purified virus obtained from allantoic fluid of 11 day-old embryonated hen's eggs was used as antigen.
Hemagglutination inhibition testHI test was performed in V-bottomed microtitre plates with 4 hemagglutinating units (HAU) of Equine Influenza virus (H3N8) antigen and 0.5% of chicken red blood cells according to standard procedures. Differences in log-transformed data from HI titers between the experimental groups were assessed using analysis of variance (ANOVA) followed by Tukey's post-hoc test. The level of statistical significance was set at P Value<0.05.
Immunoblot analysisRecombinant and viral proteins separated by SDS-PAGE were electrotransferred to a 0.22μm pore size Nitrocellulose membrane (Sigma) by using a semi-dry transfer blotter (BioRAD, Hercules, CA, USA) at 150mA for 30m. The membrane was blocked overnight at 4°C in PBS containing 5% skim milk and 0.1% Tween-20, then was reacted with 1:100 dilution of each pool of sera. After 1.5h, the membrane was washed three times with washing buffer (PBS containing 0.1% Tween-20) and subsequently incubated with horseradish peroxidase (HRP) conjugated rabbit anti-mouse IgG (Sigma) at a 1:1000 dilution for 1.5h at room temperature. After a last washing step, the membrane was developed in PBS with 0.05% diaminobenzidine tetrahydrochloride (Wako pure) in the presence of H2O2.
ResultsConstruction of the baculovirus transfer vectorRT-PCR amplification assay using HA1F and HA1R primers generated a unique fragment having the expected molecular size (1002 bp) visible on agarose gel.
After cloning the PCR product into the pCR2.1-TOPO vector and transformation in TOP10 bacteria, more than 100 clones were obtained. Twelve of them, randomly selected, were examined to ensure proper orientation by endonuclease restriction with XhoI. One clone in the correct orientation was chosen for sequence analysis. Sequencing analysis confirmed the correct open reading frame of the cloned segment. Finally, the HA1 fragment was digested with NcoI and BamHI enzymes and ligated into the pFastBac HTb vector to generate the plasmid pFastBac-HA.
Generation of the recombinant baculovirusRecombinant baculoviruses were obtained by transformation of pFastBac-HA into E. coli DH10Bac. After one and a half days of incubation in Luria broth plates, several colonies were observed. Twelve white colonies and one blue colony (non-recombinant) used as a negative control were analyzed by Colony-PCR with M13 universal primers. The transposition of the HA1 segment into the bAcNPV vector was confirmed in eleven of the apparently positive colonies analyzed.
Four days after Sf21 cell transfection, decrease of cell growth, enlargement in cell diameter and detachment of cells from the monolayer were observed. Signs of cell infection were verify 72h after re-infection of new Sf21.
Expression of the recombinant HA1 proteinAfter infection of Sf9, Sf21 and High Five cells, a recombinant protein with a molecular weight of about 40 kDa was detected by SDS-PAGE. The size of the recombinant protein was approximated to the predicted molecular weight of the HA1 containing the N terminal His tag and was also comparable to the size of the authentic viral HA1 subunit.
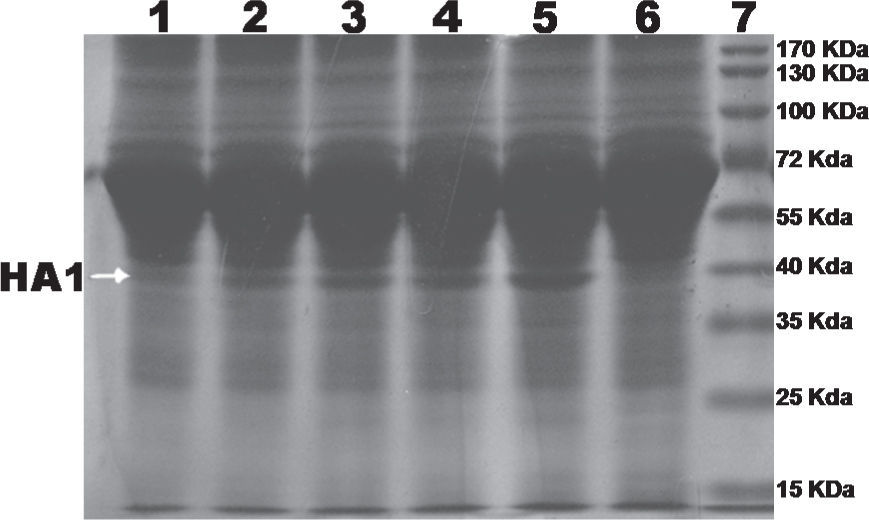
Analysis of cell lysate and cell culture media showed that the maximum expression of recombinant protein was achieved at 96 hours post-infection of High Five cells grown in TC-100 media supplemented with 10% of FBS (fig. 1). No significant expression levels were detected with Sf-9 cells or Sf-21 cells during the 96-hour analysis (data not shown).
Purification of the HA1 protein.")
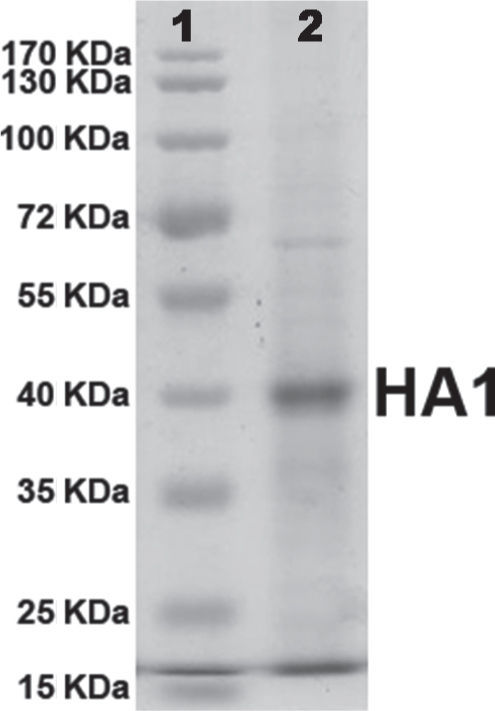
Recombinant HA1 protein was purified to near homogeneity by Immobilized Metal-ion Affinity Chromatography (IMAC) with NTA-Ni2+ resin (QIAGEN). Protein purity was analysed and confirmed by SDS-PAGE (fig. 2). Protein concentration was determined by the standard Bradford assay. After purification, a final concentration of 20μg/ml of protein was obtained.
; lane 2: purified recombinant protein by Ni2+ affinity.")
The presence of a single band on polyacrylamide gel (Fig. 2) suggested that the protein was purified by IMAC almost to homogeneity and the amount of protein quantified by the Bradford assay method corresponded completely to the recombinant HA1 protein.
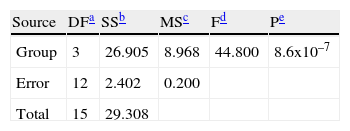
Hemagglutination inhibition testFor the statistical analysis of HI results (table 2), a logarithmic transformation of data was initially performed. No significant differences were found between the standard deviations of each group by using the Levene's test with a confidence level of 95.0% (P>0, 42). The data were analyzed by one-way Analysis of Variance (ANOVA), demonstrating significant differences between groups of inoculated animals (table 3).
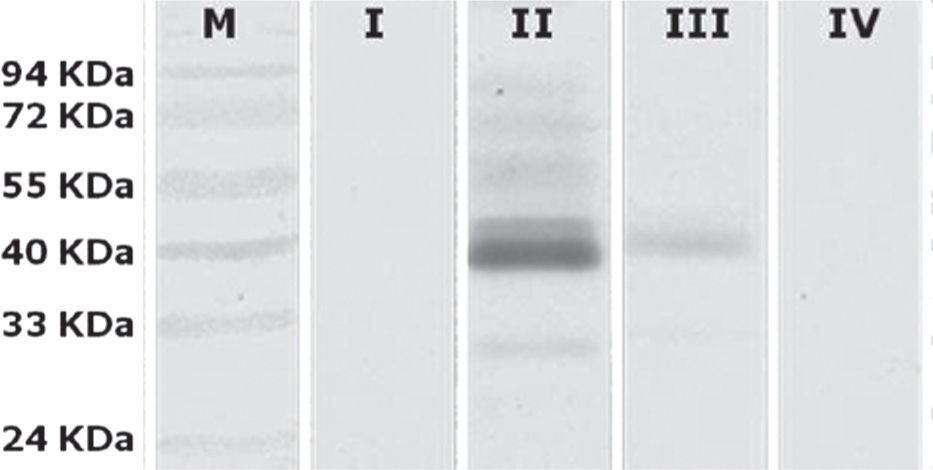
To determine which groups were significantly different, a Tukey's test was conducted. It showed that there were significant differences between Group II (animals inoculated intramuscularly with adjuvant) and other groups. Furthermore, significant differences between Group III (IM) and Group IV (not inoculated control) were found, but no observable differences were noted between Group I (IN) and control. These results were subsequently confirmed qualitatively by western blot analysis using a pool of sera from each group (fig. 3).
; lane I, Group I (intranasal); lane II, Group II (animals inoculated intramuscularly with adjuvant); lane III Group III (animals inoculated intramuscularly without adjuvant) and lane IV, Group IV (not inoculated control).")
Western blot analysis of sera pools. Lane M, PageRuler prestained marker (Fermentas); lane I, Group I (intranasal); lane II, Group II (animals inoculated intramuscularly with adjuvant); lane III Group III (animals inoculated intramuscularly without adjuvant) and lane IV, Group IV (not inoculated control).
The manufacture of current equine influenza vaccines involves the use of several batches of antigen, virus strains, cell lines, etc. that may produce differences in the quality of the immunoreactive proteins included in the vaccine. Licensed egg-grown vaccines and many cell culture-derived vaccines need bio containment facilities or harsh chemicals such as formaldehyde in the manufacturing process. The main problems for preparation of these vaccines are the large amount of embryonated eggs needed and the failure to obtain high yields of virus8. On average, the yield of total viral protein from the embryonated eggs is approximately 20-40mg per 200 eggs5. Therefore, large scale production of inactivated vaccines is relatively difficult and expensive.
The major advantage of the baculovirus expression system is that large amounts of recombinant proteins can be produced. Previous works have reported high yields of viral proteins by using baculovirus- insect cells systems. Nwe et al. obtained 77μg/106 cells of recombinant HA1 protein monolayer culture insect cells10, while Sugiura obtained 0.4–4μg/larvae of purified hemagglutinin protein from silkworm larvae infected with recombinant baculovirus14. In this study 20μg/ml (corresponding to 200μg of purified HA1 from 2.5x107 cells) were obtained from recombinant baculovirus infected cells. Since glycosylation may play an important role in the biological function of HA18-20, it is of interest to know whether the rHA produced in insect cells is properly glycosylated. During the current experiment we did not analyze the HA1 glycosylation pattern but Wang et al. have demonstrated that when a baculovirus expression system was used, all the N-linked oligosaccharide chains were present in the HA1 region indicating that the rHA produced in insect cells was indeed glycosylated with N-linked oligosaccharide side chains21. Our preliminary analysis in immunized mice indicated that the HA1 recombinant protein retained its antigenicity. In this study, Specol was used as an adjuvant. Specol was described by Bokhout et al.2. It is composed by a non-metabolisable mineral oil (Marcol 52) and two emulsifiers. Typically, Specol mainly induced antibodies of the IgG1 isotype1. Previous study in mice7 concluded that Specol might be an acceptable alternative to Freund's complete adjuvant (FCA). Specol is generally regarded as safe for use in animals1,2, showing fewer adverse effects than other wateroil adjuvants like FCA and had demonstrated strong immunostimulating properties. The use of Specol as an adjuvant significantly enhanced the immune response of immunized mice, since when administrating approximately 500ng of the protein with the adjuvant (Group II) the HI titre obtained was greater than that obtained by inoculation of 1μg of protein in aqueous solution (Group III). On the other hand, no detectable levels of antibodies were found by HI or Western blot in serum samples from animals inoculated intranasally (Group I) (Fig. 3). Previous studies reported that intranasal administration of 1.5μg of the HA recombinant baculovirus vaccine induced virus-specific antibodies, as measured by enzyme-linked immunosorbent assay (ELISA), but did not induce virus neutralizing (VN) antibodies11. This suggests that factors involving effectiveness of vaccination, such as route of administration, amount of inoculated protein, etc, may have a deep effect on the efficacy of a subunit vaccine. Taken together, the above results indicate that the HA1 subunit, expressed in insect cells as N-terminus fusion proteins with a His-Tag sequence can be produced in large quantities in a biochemically active form from a small culture volume. The protein can be easily purified, eliminating possible adverse reactions because it does not contain egg ovoalbumin. Moreover, virus inactivation or organic extraction procedures will not be necessary, thus avoiding possible denaturing effects and additional safety concerns due to residual toxic chemicals in the vaccine. Incorporating recombinant technology for the production of an equine influenza vaccine might contribute to circumvent the disadvantages of the current egg-based production method.
Although more studies are needed to assess the feasibility of the HA1 recombinant protein for vaccine production, our results demonstrate the usefulness of vaccination with an HA1 subunit in combination with an adjuvant as an approach to enhance antigen-specific immune response. Nevertheless, a suitable noninflammatory adsorptive adjuvant such as aluminum hydroxide or carbomer polymer may also be evaluated in future studies to determine the best formulation and inoculation route in horses.
The baculovirus system might be easily adapted to large scale manufacturing of different HA antigens, allowing for the development of equine influenza subunit vaccines and diagnostic tests and becoming a good alternative to the current egg-based inactivated vaccines.
Ethical disclosuresProtection of human and animal subjects. The authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of Data. The authors declare that no patient data appears in this article
Right to privacy and informed consent. The authors declare that no patient data appears in this article.
Conflicts of interestThe authors declare that they have no conflicts of interest.
This research was partially supported by Department of Science and Technology of National University of La Plata. N.A.F. hold fellowships of the Argentine Council for Scientific and Technical Research (CONICET). C.M.G. is Career Researcher of CIC-PBA.
