



The aim of the present study was to gather information regarding the molecular epidemiology of Human papillomavirus (HPV) and related risk factors in a group of women with low- and high-grade cervical lesions and cancer from the coastal region of Ecuador. In addition, we studied the evolution of HPV variants from the most prevalent types and provided a temporal framework for their emergence, which may help to trace the source of dissemination within the region. We analyzed 166 samples, including 57 CIN1, 95 CIN2/3 and 14 cancer cases. HPV detection and typing was done by PCR-sequencing (MY09/MY11). HPV variants and estimation of the time to most recent common ancestor (tMRCA) was assessed through phylogeny and coalescence analysis. HPV DNA was found in 54.4% of CIN1, 74.7% of CIN2/3 and 78.6% of cancer samples. HPV16 (38.9%) and HPV58 (19.5%) were the most prevalent types. Risk factors for the development of cervical lesions/cancer were the following: three or more pregnancies (OR=4.3), HPV infection (OR=3.7 for high-risk types; OR=3.5 for HPV16), among others. With regard to HPV evolution, HPV16 isolates belonged to lineages A (69%) and D (31%) whereas HPV58 isolates belonged only to lineage A. The period of emergence of HPV16 was in association with human populations (tMRCA=91052 years for HPV16A and 27000 years for HPV16D), whereas HPV58A preceded Homo sapiens evolution (322257 years). This study provides novel data on HPV epidemiology and evolution in Ecuador, which will be fundamental in the vaccine era.
El objetivo del presente estudio fue aportar información sobre la epidemiología molecular del virus del papiloma humano (human papillomavirus [HPV]) y los factores de riesgo asociados al desarrollo de lesiones cervicales y cáncer en mujeres de la costa del Ecuador. Además, se estudiaron la evolución de las variantes de los HPV más prevalentes y el marco temporal de su emergencia, para ayudar a rastrear la fuente de dispersión en la región. Se analizaron 166 muestras, incluyendo 57 y 95 casos de neoplasia intraepitelial cervical tipo 1 (CIN1) y tipo 2/3 (CIN2/3), respectivamente, y 14 de casos de cáncer. La detección/tipificación de HPV se realizó por PCR-secuenciación (MY09/MY11). La caracterización de variantes y la datación del ancestro común más reciente (tMRCA) se realizaron mediante filogenia y coalescencia. Se encontró ADN de HPV en el 54,4% de las muestras de CIN1, el 74,7% de las muestras de CIN2/3 y el 78,6% de las muestras de cáncer. Los tipos HPV16 (38,9%) y HPV58 (19,5%) fueron los más frecuentes. Los factores de riesgo para el desarrollo de lesiones cervicales/cáncer fueron 3 o más embarazos (OR = 4,3) e infección por HPV (O = 3,7 para HPV de alto riesgo, OR = 3,5 para HPV16), entre otros. En cuanto a la evolución viral, los aislados del HPV16 pertenecían a los linajes A (69%) y D (31%), mientras que los aislados del HPV58 pertenecían únicamente al linaje A. El período de emergencia del HPV16 estuvo asociado a poblaciones humanas (tMRCA = 91.052 años para HPV16A y 27.000 para HPV16D), mientras que el del HPV58A precedió a la evolución de Homo sapiens (322.257 años). Este estudio proporciona datos novedosos sobre la epidemiología y la evolución del HPV en Ecuador, los cuales serán fundamentales en la era de la vacuna.
For more than 20 years, worldwide epidemiological studies have established the association between Human papillomavirus (HPV) genital infection and the development of cervical cancer. Such association is strong for some oncogenic viral types (“high-risk types”) which have been categorized as human carcinogens by the International Agency for Research on Cancer (IARC)29. HPVs which are clinically relevant belong to the family Papillomaviridae, genus Alphapapillomavirus, species A5 (HPV51), A6 (HPV56), A7 (HPV18, 39, 45, 59) and A9 (HPV16, 31, 33, 35, 52, 58)22,38. Moreover, the genetic variation within a viral type has been recognized as an additional risk factor for cervical cancer progression3,6,9,44. These “genetic variants” are isolates of the same type that vary by less than 2% in their genome sequences and can be classified with a phylogenetically-based taxonomy9,22,44. These markers have been poorly studied in Ecuador, with only two previous reports34,47.
In Ecuador, cervical cancer is the first most common cancer among women at reproductive age (15–44 years) with an estimate of 2094 new cases and 1026 deaths in 20148. The highest incidence rates are observed in the 40–64 year-age group8. In addition, the national Ecuadorian registry showed that the incidence rates are not homogeneous throughout the country, with the highest incidence rates observed in the city of Cuenca (31.3/100000 women) and the lowest in Quito (17.7/100000 women)41. Therefore, screening for cervical cancer and HPV infection remains an important health concern throughout the country.
During the last 20 years, very few studies have partially addressed this issue, with diverse findings4,7,10,12,13,25,27,34,39,47. For instance, some studies have shown that HPV prevalence among women with normal cytology ranges from 5% to 50.5%12,13, with the most prevalent types being HPV5112 and HPV6610. With respect to abnormal cytology, other studies have shown molecular evidence indicating that HPV16 is the most prevalent type34,39, with the exception of one study, in which HPV18 was the most prevalent one (15%)4. These differences across studies might be explained by variations in the population and/or in the methods used. For example, many studies did not perform the genotyping for a broad range of HPV types39, and the cyto-histological diagnosis of samples was not always clear in the data7,10,12,25,27. Altogether, there are no data available from Ecuador regarding HPV prevalence in the Institut Català d’Oncologia (ICO) Information Centre on HPV and Cancer8.
In this context, the aim of the present study was to gather information regarding the molecular epidemiology of HPV and related risk factors in a group of women with low- and high-grade cervical lesions and cancer from the coastal region of Ecuador. In addition, we studied the evolution of HPV variants from the most prevalent types and provided a temporal framework for their emergence, which may help to trace the source of dissemination within the region.
Materials and methodsStudy design and general populationWomen diagnosed with cervical lesions and cancer attending several community health centers affiliated or not to the Sociedad de Lucha Contra el Cancer de Ecuador (SOLCA), in the provinces of Esmeraldas, Manabí, Los Ríos, Santa Elena, Guayas and El Oro (coastal region of Ecuador) were informed about the project and invited to participate during 2014.
A standardized questionnaire was administered to each participant in order to characterize the population. Items included questions regarding economic status, education, sexual and reproductive history (menarche, age at first sexual intercourse, number of sexual partners in the last two years, use of oral contraceptives (OCs), number of pregnancies), and history of sexually transmitted diseases (STDs).
Ethics, consent and permissionsThe research protocol of the study was reviewed and approved by the Research Board of Hospital del Niño Francisco de Icaza Bustamante, Guayaquil, Ecuador (Date 04/12/2013; no reference number). All procedures were in accordance with the Helsinki Declaration. Data confidentiality was maintained throughout the study.
Biological samplesCervical swabs and/or fresh biopsies were obtained by trained gynecologists at each healthcare center. Samples were collected in Cobas PCR cell Collection medium (Roche Molecular Systems) and sent to the facilities of Instituto Nacional de Investigación en Salud Pública and Instituto de Biomedicina, Faculty of Medicine, Universidad Católica de Santiago de Guayaquil, Guayaquil, Ecuador for HPV-DNA analysis. Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen) and quantified using NanoDrop 2000 (ThermoScientific™). Subsequently, DNA was subjected to PCR amplification of 110bp of the human beta-globin gene for quality control with primers PC03 5′-ACACAACTGTGTTCACTAGC-3′ and PC04 5′CAACTTCATCCACGTTCACC-3′.
NomenclatureSamples were classified according to the histological type as: Cervical Intraepithelial Neoplasia types 1, 2 and 3 (CIN1, CIN2 and CIN3 respectively) and cancer (invasive carcinoma and adenocarcinoma). Although CIN2 and CIN3 are considered intraepithelial lesions endowed with distinct risks of persistence (35% and 56% respectively) and progression toward invasion (5% and >12% respectively)48, the CIN2 is utilized as a threshold for treatment in clinical practice48,49. Thus, our study has adhered to combine CIN2/3 as a single category to enhance statistical power by forming a high-grade class of clinical meaning (see statistical analysis).
HPV variants were classified into a phylogenetically-based taxonomy of lineages and sub-lineages following the alphanumeric system described by Burk et al.9 According to this, HPV16 can be classified into four lineages: A (formerly European), B (African type 1), C (African type 2) and D (Asian American) an nine sub-lineages (A1, A2, A3, A4, B1, B2, D1, D2, D3), whereas HPV58 can be classified into four lineages (A, B, C, D) with seven sub-lineages (A1, A2, A3, B1, B2, D1, D2)9.
Study population sizeA total of 201 women provided informed consent to participate in this study. Thirty five (35) out of a total of 201 collected samples had to be excluded due to problems in the quality of the material (negative for the human beta-globin gene). The present analysis refers to the remaining 166 evaluable samples, including 57 with CIN1, 95 CIN2/3 and 14 cancer cases.
HPV DNA analysisHPV detection was performed with L1 consensus primers MY09-MY1132. Briefly, PCR products were visualized by 2% agarose gel electrophoresis with SYBR Safe (Life Technologies) staining. Positive amplicons were purified with the QIA quick Gel Extraction kit (Qiagen). In order to obtain DNA sequences, amplicons were submitted, along with the original forward and reverse primers to Genewiz Inc. This sequencing service was not involved in the study design or data analysis (sampling, DNA extraction, HPV-PCR detection, bioinformatics for sequence typing, phylogeny). However, it provides a series of .abi, .txt and .pdfs files from each sample.
The L1- MY09-MY11 sequences were read and analyzed in our lab using Codon Code aligner software v. 3.0.1 (Codon Code Corporation). The first 20 nucleotides of each strand were trimmed to exclude illegible regions. The samples that were unequivocally aligned at both strands were considered to be single infections, while those with readable chromatograms but containing several positions with double peaks were considered an infection with multiple genotypes. In order to provide a potential combination of types, several chromatograms per each sample were analyzed. In most cases, one type showed the best signal sequence over another, allowing its identification. In other cases, the identification was partial, and the potential types were left as “undetermined” (HPV?). HPV types were defined based on sequence identity using Blast search2.
HPV16 and 58 phylogenetic analysisFor HPV16, the obtained sequences were classified based on phylogenetic analysis as lineages A, B, C or D by using the following reference sequences from Burk et al.9, A1 (K02718), A2 (AF536179), A3 (HQ644236), A4 (AF534061), B1 (AF536180), B2 (HQ644298), C (AF472509), D1 (HQ644257), D2 (AY686579), D3 (AF402678). Phylogenetic trees were constructed using sequences from this study (n=35), 36 published sequences from Ecuador34,47 and 53 worldwide reference lineages9,44.
For HPV58, the obtained sequences were classified as lineages A, B, C and D. Phylogenetic trees were constructed using the following reference sequences from Burk et al.9, A1 (D90400), A2 (HQ537752), A3 (HQ537758), B1 (HQ537762), B2 (HQ537764), C (HQ537774), D1 (HQ537768), D2 (HQ537770), worldwide lineages from Chan et al.14 (n=45) and Ecuadorian sequences from this study (n=15) and Mejia et al.34 (n=12). The two phylogenetic trees, scaled in calendar dates, were obtained using the Bayesian method implemented in the BEAST v 1.8.3 software24.
Coalescent analysisEstimation of the time to most recent common ancestor (tMRCA) of the sequences of HPV16 and HPV58 was carried out by Monte Carlo Markov Chain (MCMC) Bayesian coalescent analysis implemented in BEAST v 1.8.324. To select the nucleotide substitution model that best fits the sequence data, we used jModelTest v 2.1.320 and the Akaike Information Criterion (AIC). For HPV58, the selected model was GTR. For HPV16, the selected one was TPM1uf: Kimura 81 with unequal base frequencies. The Bayesian Skyline Plot (BSP) was selected as a model to estimate the evolutionary and coalescent parameters23. BSPs were run under the two molecular clock models – strict and relaxed uncorrelated lognormal. A substitution rate of 1.84×10−8 substitutions per site per year (s/s/y) was set according to Chen et al.17 This theoretical value comes from fossil calibration points for the Felidae Papillomavirus tree40. The best clock model (strict) was chosen based on a Bayesian Factor analysis.
MCMC were run for 5×107 generations, sampling every 5000th generation in order to achieve an Effective Sample Size (ESS)>200. All BEAST run logs were analyzed with the TRACER program version 1.5 (http://beast.bio.ed.ac.uk/treeannotator) after discarding 2% of the run length as burn-in. The maximum clade credibility tree (MCCT) was constructed with the Tree Annotator tool (http://beast.bio.ed.ac.uk/treeannotator) after discarding 2% of the sampling. The MCCT summarizing the posterior information of topologies and the median branch lengths from the trees sampled was then visualized with FigTree V1.4.0 software (http://tree.bio.ed.ac.uk/).
Statistical analysisData are presented as frequencies and percentages. The distribution of HPV types and/or socio-cultural variables according to lesion grade was compared by χ2 or the two-tailed Fisher exact test. Logistic regression was used to estimate the odds ratio (OR) and 95% confidence intervals (CIs) using SPSS software (SPSS, Inc., Chicago, USA).
Statistical analysis was done by combining histological results into three categories, CIN1, CIN2/3 and cancer. OR estimates were based on bivariate categories using CIN1 as reference (i.e., CIN1 vs. CIN2/3 and CIN1 vs. cancer).
GenBank accession numberA total of 88 sequences of ∼400bp, which belong to HPV6, 11, 16, 18, 31, 33, 35, 42, 51, 52, 53, 56, 58, 61, 62 and 70 were deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession numbers: [KU050106–KU050193].
ResultsStudy populationAccording to our survey, 75.3% of all interviewed women were unemployed (housewives) and nearly half of the study population had not achieved high school education or higher. The median age of menarche and first sexual intercourse was 13 and 17 years respectively and a substantial proportion (58.5%) had had multiple pregnancies, with a median of four. The general characteristics of the study population are summarized in Table S1.
HPV DNA analysisA hundred and thirteen (113) samples were positive for HPV (68.1%). Ninety-three (82.3%, 93/113) samples were single infections whereas 11.5% (13/113) were identified as multiple infections. After stratifying by histological type, HPV DNA was found in 54.4% (31/57) of CIN1, 74.7% (71/95) of CIN2/3 and 78.6% (11/14) of the cancer samples.
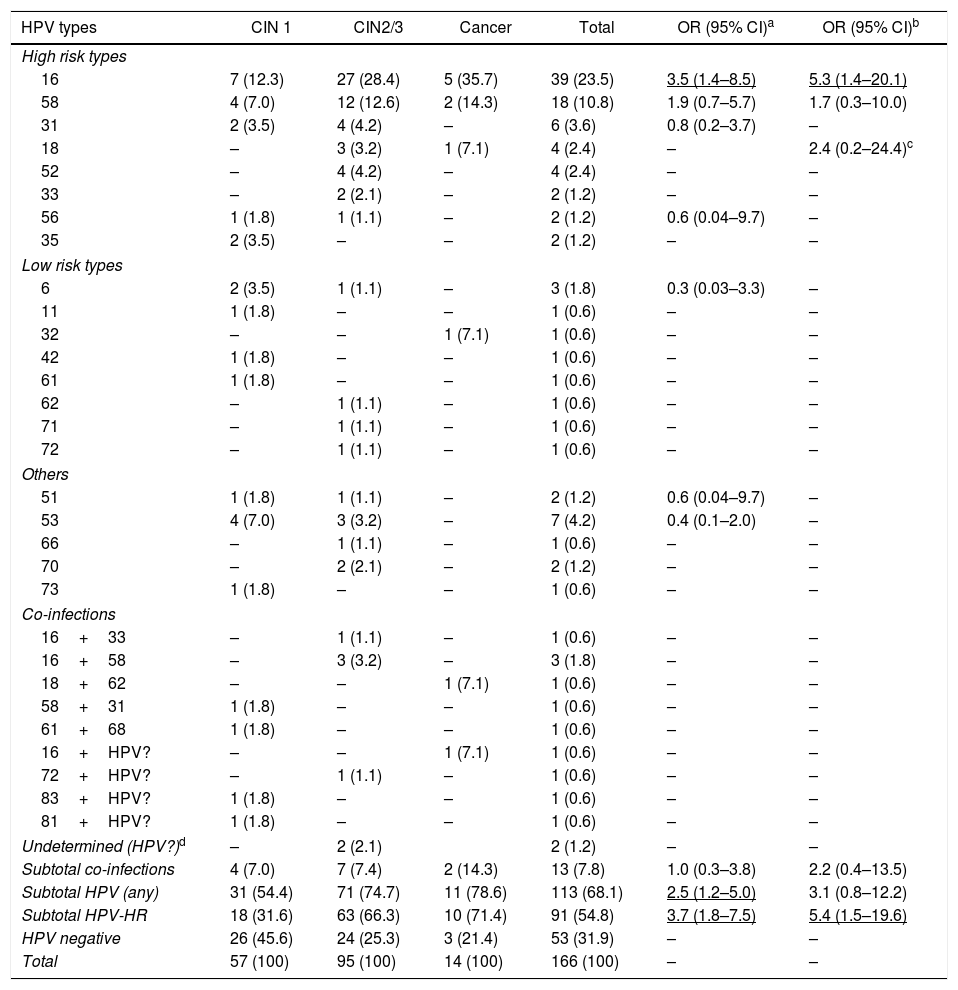
Among the positive samples, the most common viral types were HPV16, with 38.9% (44/113), and HPV58, with 19.5% (22/113). HPV18 was the fourth most common viral type. Details of HPV types stratified by Pap diagnosis are shown in Table 1.
Distribution of HPV types according to histological type
| HPV types | CIN 1 | CIN2/3 | Cancer | Total | OR (95% CI)a | OR (95% CI)b |
|---|---|---|---|---|---|---|
| High risk types | ||||||
| 16 | 7 (12.3) | 27 (28.4) | 5 (35.7) | 39 (23.5) | 3.5 (1.4–8.5) | 5.3 (1.4–20.1) |
| 58 | 4 (7.0) | 12 (12.6) | 2 (14.3) | 18 (10.8) | 1.9 (0.7–5.7) | 1.7 (0.3–10.0) |
| 31 | 2 (3.5) | 4 (4.2) | – | 6 (3.6) | 0.8 (0.2–3.7) | – |
| 18 | – | 3 (3.2) | 1 (7.1) | 4 (2.4) | – | 2.4 (0.2–24.4)c |
| 52 | – | 4 (4.2) | – | 4 (2.4) | – | – |
| 33 | – | 2 (2.1) | – | 2 (1.2) | – | – |
| 56 | 1 (1.8) | 1 (1.1) | – | 2 (1.2) | 0.6 (0.04–9.7) | – |
| 35 | 2 (3.5) | – | – | 2 (1.2) | – | – |
| Low risk types | ||||||
| 6 | 2 (3.5) | 1 (1.1) | – | 3 (1.8) | 0.3 (0.03–3.3) | – |
| 11 | 1 (1.8) | – | – | 1 (0.6) | – | – |
| 32 | – | – | 1 (7.1) | 1 (0.6) | – | – |
| 42 | 1 (1.8) | – | – | 1 (0.6) | – | – |
| 61 | 1 (1.8) | – | – | 1 (0.6) | – | – |
| 62 | – | 1 (1.1) | – | 1 (0.6) | – | – |
| 71 | – | 1 (1.1) | – | 1 (0.6) | – | – |
| 72 | – | 1 (1.1) | – | 1 (0.6) | – | – |
| Others | ||||||
| 51 | 1 (1.8) | 1 (1.1) | – | 2 (1.2) | 0.6 (0.04–9.7) | – |
| 53 | 4 (7.0) | 3 (3.2) | – | 7 (4.2) | 0.4 (0.1–2.0) | – |
| 66 | – | 1 (1.1) | – | 1 (0.6) | – | – |
| 70 | – | 2 (2.1) | – | 2 (1.2) | – | – |
| 73 | 1 (1.8) | – | – | 1 (0.6) | – | – |
| Co-infections | ||||||
| 16+33 | – | 1 (1.1) | – | 1 (0.6) | – | – |
| 16+58 | – | 3 (3.2) | – | 3 (1.8) | – | – |
| 18+62 | – | – | 1 (7.1) | 1 (0.6) | – | – |
| 58+31 | 1 (1.8) | – | – | 1 (0.6) | – | – |
| 61+68 | 1 (1.8) | – | – | 1 (0.6) | – | – |
| 16+HPV? | – | – | 1 (7.1) | 1 (0.6) | – | – |
| 72+HPV? | – | 1 (1.1) | – | 1 (0.6) | – | – |
| 83+HPV? | 1 (1.8) | – | – | 1 (0.6) | – | – |
| 81+HPV? | 1 (1.8) | – | – | 1 (0.6) | – | – |
| Undetermined (HPV?)d | – | 2 (2.1) | 2 (1.2) | – | – | |
| Subtotal co-infections | 4 (7.0) | 7 (7.4) | 2 (14.3) | 13 (7.8) | 1.0 (0.3–3.8) | 2.2 (0.4–13.5) |
| Subtotal HPV (any) | 31 (54.4) | 71 (74.7) | 11 (78.6) | 113 (68.1) | 2.5 (1.2–5.0) | 3.1 (0.8–12.2) |
| Subtotal HPV-HR | 18 (31.6) | 63 (66.3) | 10 (71.4) | 91 (54.8) | 3.7 (1.8–7.5) | 5.4 (1.5–19.6) |
| HPV negative | 26 (45.6) | 24 (25.3) | 3 (21.4) | 53 (31.9) | – | – |
| Total | 57 (100) | 95 (100) | 14 (100) | 166 (100) | – | – |
Values are expressed as n (%); significant p values (p<0.05) are underlined.
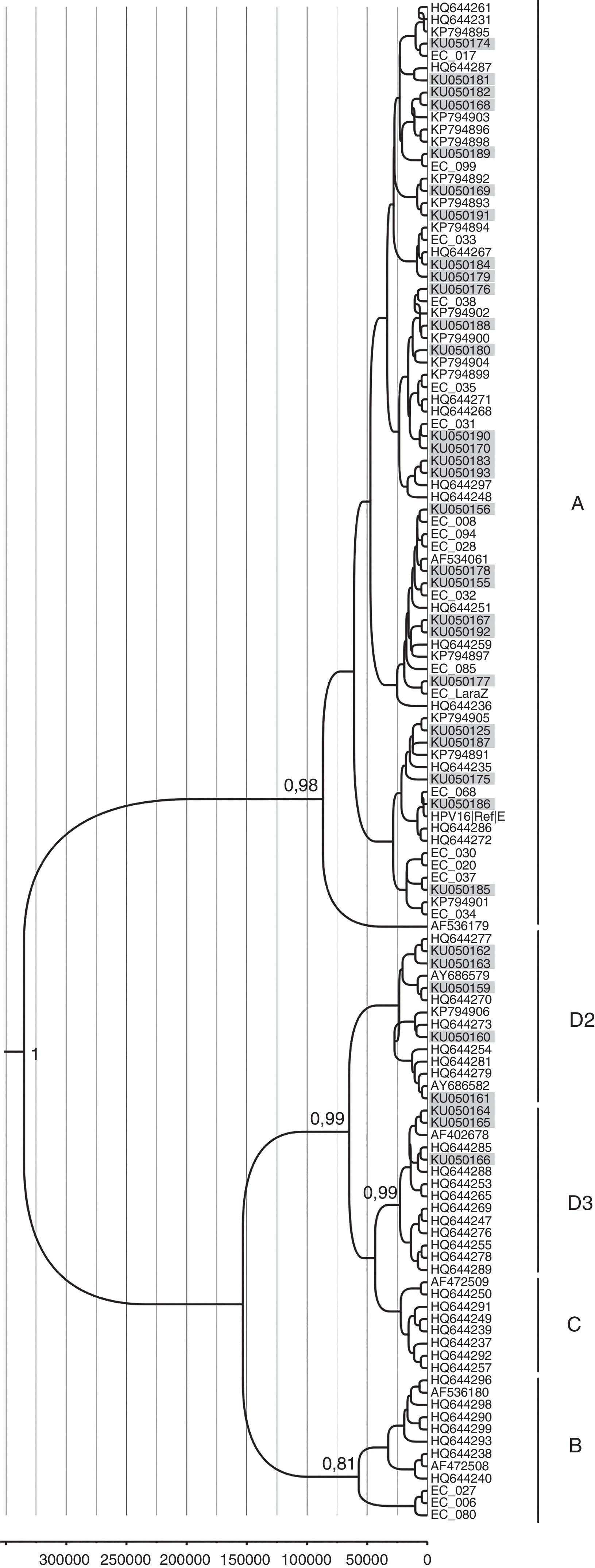
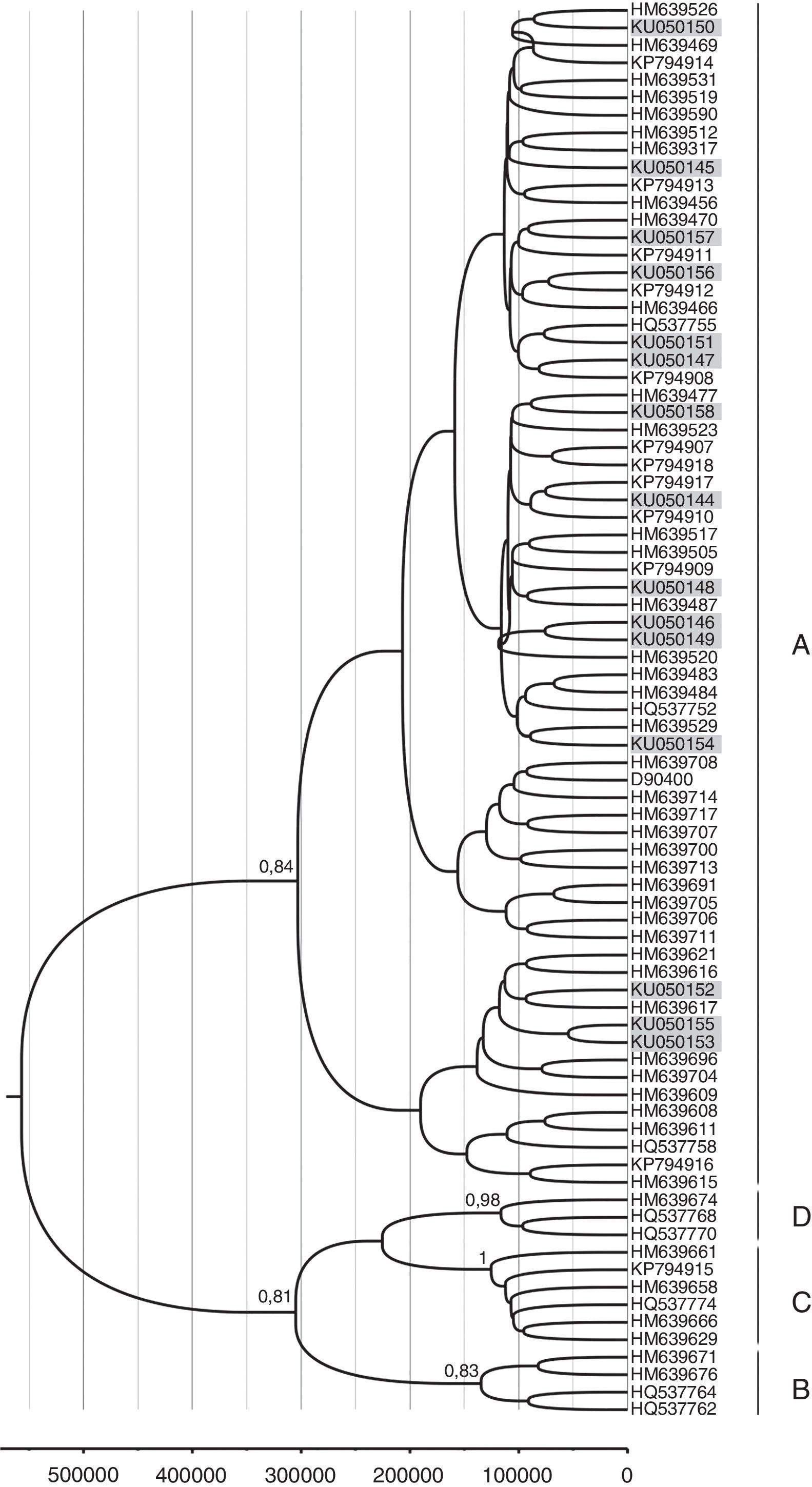
A total of 42 HPV16 sequences and 15 HPV58 sequences were adequate for phylogeny (>400bp sequence length). The phylogenetic analysis of HPV16 variants showed that 69% (29/42) of the isolates clustered with lineage A and 31% (13/42) with lineage D. Among the D isolates, HPV16 variants belonged to sub-lineages D2 and D3, with 46.1% (6/13) and 53.9% (7/13) respectively (Fig. 1). Moreover, all HPV58 sequences belonged to lineage A, sub-lineages A2 with 80% (12/15) and A3 with 20% (3/15) (Fig. 2).
 are shown next to the branches.")
Phylogenetic analysis and molecular dating of HPV-16 variants from this study. The evolutionary history of HPV-16 variants was inferred using the Bayesian method. The maximum clade credibility tree is shown. Timeline: the X axis indicates years ago. The posterior probability values (p=0.89) are shown next to the branches.
 are shown next to the branches.")
Phylogenetic analysis and molecular dating of HPV-58 variants from this study. The evolutionary history of HPV-58 variants was inferred using the Bayesian method. The maximum clade credibility tree is shown. Timeline: the X axis indicates years ago. The posterior probability values (p=0.89) are shown next to the branches.
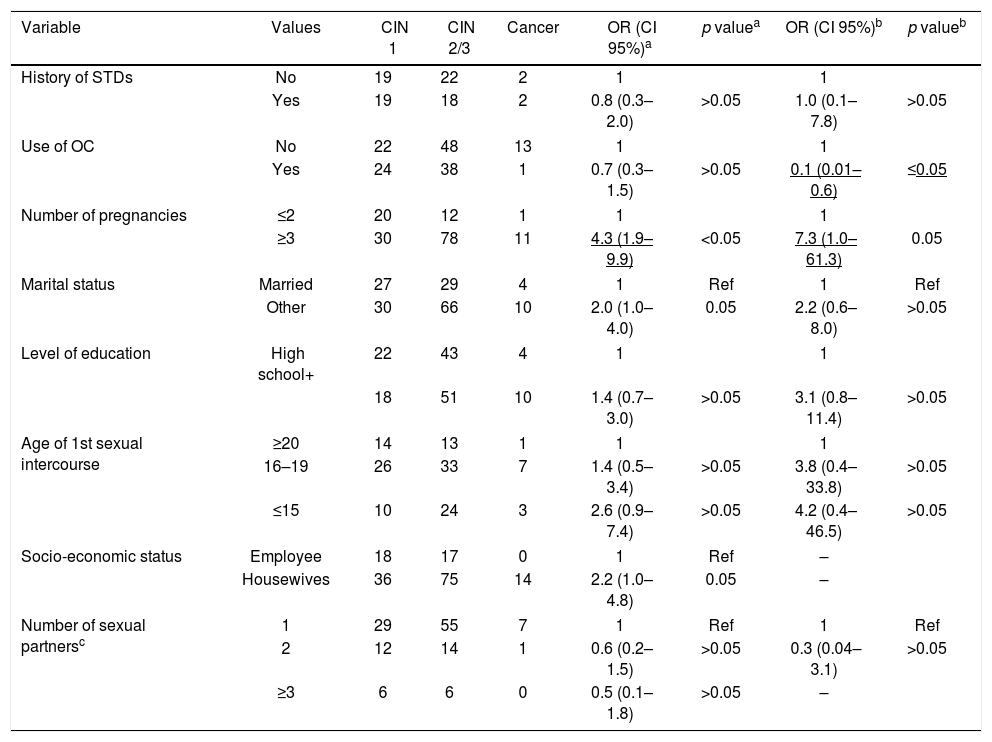
Risk factors related to the development of CIN2/3 determined by logistic regression were: HPV infection with an OR of 2.5 (1.2–5.0), HPV type with an OR of 3.7 (1.8–7.5) for any high-risk HPV type and OR of 3.5 (1.4–8.5) for HPV16, number of pregnancies with an OR of 4.3 (1.9–9.9) for three or more pregnancies. Risk factors related to the development of cancer were: HPV type with an OR of 5.4 (1.5–19.6) for any high-risk HPV type and OR of 5.3 (1.4–20.1) for HPV16. Results are shown in Table 2. The distribution of HPV lineages among cervical lesions was not statistically significant, although the number of isolates was too small to be conclusive. Details on HPV variants by histological results are shown in Table S2.
Socio-cultural risk factors evaluated in relation to cervical lesions and cancer development
| Variable | Values | CIN 1 | CIN 2/3 | Cancer | OR (CI 95%)a | p valuea | OR (CI 95%)b | p valueb |
|---|---|---|---|---|---|---|---|---|
| History of STDs | No | 19 | 22 | 2 | 1 | 1 | ||
| Yes | 19 | 18 | 2 | 0.8 (0.3–2.0) | >0.05 | 1.0 (0.1–7.8) | >0.05 | |
| Use of OC | No | 22 | 48 | 13 | 1 | 1 | ||
| Yes | 24 | 38 | 1 | 0.7 (0.3–1.5) | >0.05 | 0.1 (0.01–0.6) | ≤0.05 | |
| Number of pregnancies | ≤2 | 20 | 12 | 1 | 1 | 1 | ||
| ≥3 | 30 | 78 | 11 | 4.3 (1.9–9.9) | <0.05 | 7.3 (1.0–61.3) | 0.05 | |
| Marital status | Married | 27 | 29 | 4 | 1 | Ref | 1 | Ref |
| Other | 30 | 66 | 10 | 2.0 (1.0–4.0) | 0.05 | 2.2 (0.6–8.0) | >0.05 | |
| Level of education | High school+ | 22 | 43 | 4 | 1 | 1 | ||
| 18 | 51 | 10 | 1.4 (0.7–3.0) | >0.05 | 3.1 (0.8–11.4) | >0.05 | ||
| Age of 1st sexual intercourse | ≥20 | 14 | 13 | 1 | 1 | 1 | ||
| 16–19 | 26 | 33 | 7 | 1.4 (0.5–3.4) | >0.05 | 3.8 (0.4–33.8) | >0.05 | |
| ≤15 | 10 | 24 | 3 | 2.6 (0.9–7.4) | >0.05 | 4.2 (0.4–46.5) | >0.05 | |
| Socio-economic status | Employee | 18 | 17 | 0 | 1 | Ref | – | |
| Housewives | 36 | 75 | 14 | 2.2 (1.0–4.8) | 0.05 | – | ||
| Number of sexual partnersc | 1 | 29 | 55 | 7 | 1 | Ref | 1 | Ref |
| 2 | 12 | 14 | 1 | 0.6 (0.2–1.5) | >0.05 | 0.3 (0.04–3.1) | >0.05 | |
| ≥3 | 6 | 6 | 0 | 0.5 (0.1–1.8) | >0.05 | – | ||
Statistically significant associations are underlined (p<0.05).
The molecular dating of HPV16 and HPV58 phylogenetic trees is shown as a ruler at the bottom of each Figures 1 and 2. The mean estimates for the tMCRA supported with a posterior value>0.8 were as follows: HPV16 root=339353 (HPD 95%=111788–670346); HPV16D3=27046 (HPD 95%=5323–52590); HPV16D2=27817 (HPD 95%=6198–55704); HPV16A=91052 (HPD 95%=19529–230565). HPV58 root=576059 (HPD 95%=245533–1016441); HPV58A=322257 (HPD 95%=139260–562715); HPV58B=153664 (HPD 95%=61227–265684); HPV58C=144449 (HPD 95%=65850–216097); HPV58D=135470 (HPD 95%=57266–207732).
DiscussionThe perspectives of administration of prophylactic vaccines highlight the need to reinforce the knowledge of the type-specific prevalence of high-risk HPVs in Ecuador. This study is among the largest conducted to date on HPV type distribution in Ecuadorian women with cervical lesions and the only one to show recorded data on demographic, gynecological and socio-cultural characteristics of the population.
HPV DNA was found in 54.4% (31/57) of CIN1, 74.7% (71/95) of CIN2/3 and 78.6% (11/14) of cancer samples. Although these values seem to be low for CIN, they are in the range of those previously reported for Ecuador in similar studies7,13,25,27,34,39,47. Nevertheless, since the MY09/11+sequencing system has been reported to be less efficient to detect multiple infections compared to other methods5, we cannot rule out a potential underestimation of the type-specific prevalence, which may be a limitation of this study.
Risk factors related to the presence of CIN2/3 and cancer detected in the present study were: HPV infection and number of pregnancies, which is in agreement with some previous studies30,37. Cervical carcinogenesis appears to involve high levels of sex hormones and estrogens have been suggested to stimulate HPV gene expression, influence the cervical immune response, and stimulate cell proliferation in the transformation zone21. Exposure to sex hormones is related both to parity and to OC use. However, we found no increased risk for CIN2/3 or cancer in relation to OC use. A possible explanation for this is that pregnancy is characterized by intense short-term exposure to hormones, whereas OC use is characterized by low-intensity exposure and can often need longer exposure to become a risk factor (>5 years)36. The absence of data on the duration of OC use in our population limited our appropriate evaluation of the latter variable.
In our study population, HPV16 was the most common oncogenic type (38.9% of positive samples). Women infected by HPV16 were about five times more likely to develop cervical cancer than their counterparts infected by any other types. The prevalence of HPV16 type is in accordance with worldwide data19 and most local Ecuadorian studies34,39.
Our data also showed considerable frequencies of HPV58 (19.5%), a type that accounts for a notable proportion of cervical cancers in East Asia (∼50%) but for less than 10% in South America19,33. In agreement with our data, this local trend in HPV58 prevalence is supported by a recently published study carried out in the main Ecuadorian city of Quito, in which HPV58 shows a prevalence of 30.5%34.
The Costa Rica Vaccine Trial using the bivalent HPV16/18 vaccine has recently demonstrated partial cross-protection against HPV31, HPV33 and HPV45, and increased HPV51 incidence28. It will be necessary to continue the surveillance of HPV58 prevalence as immunization programs are introduced to assess a potential scenario of viral replacement in high-grade squamous intraepithelial lesion (HSIL) and cancer in Ecuador.
The phylogenetic analysis of HPV16 variants showed that 68% of HPV16 variants belong to lineage A and 31% to lineage D. From a clinical point of view, it is known that lineage D variants are associated with an increased risk of persistent infection and development of cervical lesions in Latin American women3,6,9,44. Although in the present study the number of isolates was too small to conduct a statistically significant analysis, the proportion of those variants was higher in cancer (in situ and invasive) (40%) than in CIN1 (28%) cases. Moreover, the prevalence of HPV16 lineage D reported here was higher than that reported in other South American countries such as Paraguay (15%) and Argentina (10%), but similar to that reported for the border country of Colombia (33%)3,31,35. These differences may be related to the ethnic composition of the studied populations, but also to migration patterns among countries, thus highlighting the large influx of Colombians and Peruvians to Ecuador during the last decades42.
With regard to HPV58, our phylogenetic analysis indicated that 80% belonged to lineage A2 and 20% to lineage A3. The latter has been reported as more oncogenic than other lineages15 and is frequent in Asia (34%) and the Americas (11%) but accounts for less than 5% in Europe and Africa14.
The molecular dating of HPV lineages within each type may allow us to hypothesize about the circulation of those isolates in Ecuador. In this study, all variants diverged from their common ancestors within the last million years (HPV16 mean=339353ybp and HPV58 mean=576059ybp), but the time of emergence of some sub-lineages (such as the aggressive variants HPV16 D3 and D2) is a more recent event, estimated to be nearly 28000 years ago. During that time, most human populations were fully geographically established on earth and therefore, those dates of emergence may explain the ethnogeographical patterns associated with Amerindian populations1,46.
Under this scenario, the current patterns of HPV16 infection are the result of a combination of dispersal events (migration and founder effects) and viral co-evolution with humans. In Ecuador, genetic admixture measured with autosomal loci is 73% Native-American, 19% European and 8% African, whereas that measured at the Y chromosome level is 70% European, 28% Amerindian, and 2% African26. Thus, it is possible that the described proportions of infections with HPV16 variants reflect the paternal side of the admixture process during the conquest, which frequently involved European males and Amerindian women43.
Conversely, for HPV58, the divergence of each sub-lineage can be estimated in a time frame before the date on which modern humans expanded out of Africa (>100000ybp). Our estimates are also in accordance with those published by Marin et al.33 for HPV58 in Argentina. Under this scenario, the current patterns of viral infection are the result of main dispersal events rather than of co-evolution with those populations. The fact that the tree of HPV58 does not reflect human evolution to the extent shown by HPV16 has been previously observed by other authors11,16,18,45, but to the best of our knowledge, this is the first time that a time frame for that observation is provided. A potential limitation of this analysis is further assessed in Supplementary Text 3.
Overall, this study contributes at building our knowledge related to the epidemiology of cervical cancer in Ecuadorian women with low- and high-risk cervical lesions. Before this study, only 28 sequences of HPV L1 from Ecuador were available at Genbank. This work will significant enlarge that dataset by adding 88 sequences. This information will be fundamental for local decision-makers to consider cervical cancer screening programs and useful globally for the understanding of HPV variant distribution around the world.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors must have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence must be in possession of this document.
FundingThis work was funded by Secretaria Educación Superior, Ciencia, Tecnología e Innovación (SENESCYT) from Ecuador (agreement 20150044 CI) and Proyecto Prometeo (SENESCYT, 2015).
Conflict of interestThe authors declare that they have no conflicts of interest.
The authors would like to thank the healthcare professionals participating at the multiple centers who aided at collecting clinical data and samples, such as Hospital de Infectología Dr. Jose Rodriguez Maridueña, Hospital Gineco-Obstétrico Enrique C. Sotomayor, SOLCAs from Guayaquil, Portoviejo, Manta, Esmeraldas, Machala, Santa Elena, Hospital Liborio Panchana from Santa Elena, Hospital MaternoInfantil “Enrique Ponce Luque” from Babahoyo, Direcciones Regionales de MSP zona 5 & 8, Distrito de Salud 12 D01-Los Ríos. In addition, we thank professionals at several laboratories who aided at primarily processing many of the samples: Laboratorio de Biomedicina ESPOL, Instituto de Biomedicina, Universidad Católica de Santiago de Guayaquil, Laboratorio de Virologíadel IPK.
The following are the supplementary data to this article:
