



Two cross-sectional studies were carried out in 2013 and 2015 monitoring for Mycoplasma hyopneumoniae presence in a swine farm. In these studies, the genetic diversity of M. hyopneumoniae was assessed in clinical specimens using a Multiple Locus Variable-number tandem repeat Analysis (MLVA) targeting P97 R1, P146 R3 and H4 loci. The samples from August 2015 showed the MLVA profile prevalent in June 2013, therefore it can be concluded that a same genetic type of M. hyopneumoniae can persist for at least two years in a closed herd. In addition, the nested PCR reactions implemented in this study showed to be useful for MLVA typing in non-invasive clinical samples.
Dos estudios transversales fueron realizados en los años 2013 y 2015 monitorizando la presencia de Mycoplasma hyopneumoniae en una piara. En esos estudios la diversidad genética de M. hyopneumoniae fue evaluada a partir de muestras clínicas utilizando un análisis multilocus de regiones repetidas en tándem (MLVA) de los loci P97 R1, P146 R3 y H4. Las muestras colectadas en agosto del 2015 mostraron el perfil de MLVA prevalente en junio del 2013, por lo tanto, se puede concluir que el mismo tipo genético de M. hyopneumoniae puede persistir por al menos 2 años en una piara sin reposición externa de animales. Además, las reacciones de PCR anidadas implementadas en este estudio mostraron ser útiles para la tipificación por MLVA a partir de muestras clínicas no invasivas.
Mycoplasma hyopneumoniae is the etiological agent of enzootic pneumonia, an important and highly prevalent respiratory disease affecting pigs in almost all swine-producing areas around the world. Certain epidemiological aspects, such as persistence and genetic diversity of M. hyopneumoniae strains circulating within herds, have been addressed in some studies9,11, however, most of the longitudinal studies were carried out over a period of less than one year.
Even though there is no molecular marker able to discriminate between high and low virulence M. hyopneumoniae strains, the knowledge of the persistence of certain genotypes of the agent might be an important indicator of the severity of the disease when comparing clinical signs and lung lesions.
The aim of this study was to determine genotypes of M. hyopneumoniae present in pigs in a commercial farm on two sampling occasions, two years apart, to assess persistence of the strains.
The study was performed according to the international guidelines of the Council for International Organizations of Medical Sciences (CIOMS).
Typing of M. hyopneumoniae was performed by Multiple-Locus Variable-Number Tandem-Repeat Analysis (MLVA) using DNA samples positive for M. hyopneumoniae by a nested PCR (nPCR) described by Calsamiglia et al.1 P97 R1, P146 R3 and H4 loci were analyzed in 35 DNA samples obtained from nasal swab (NS) and bronchoalveolar lavage (BAL) specimens of sows and pigs. All samples were collected in two cross-sectional studies performed on June 2013 (27 specimens) and on August 2015 (8 specimens) in order to monitor enzootic pneumonia control measures in a two-site, 1300-sow, commercial wean-to-finish farm. The control measures applied since June 2013 included sows vaccination against M. hyopneumoniae (Porcilis M hyo, MSD Animal Health) and strategic in-feed antibiotic treatment (tiamulin at 112ppm and chlortetracycline at 300ppm) to pigs at 6 weeks of age during a 2-week period.
For MLVA, VNTR loci P97 R1 and H4 were amplified by nPCR format combining primers designed in previous studies. The primers reported by de Castro et al.2, were used for the first round of amplification, and those reported by Vranckx et al.10, for the second round. The P146 R3 locus was amplified using a nPCR previously reported by Tamiozzo et al.8,9 After amplification, the amplicons were purified (Puriprep-GP Kit, Inbio Highway), quantified and sequenced (ABI 3130xl; Applied Biosystems) with the primers described by Vranckx et al.10, for P97 and H4, and those of Mayor et al.6, for P146 R3. The number of tandem repeats was determined viewing the sequences in the BioEdit software.
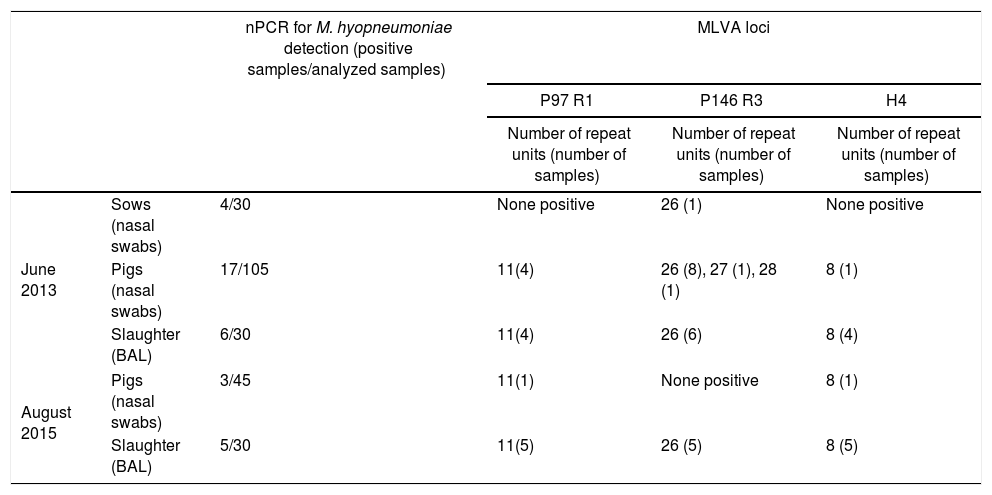
All the samples from June 2013 and August 2015 characterized by MLVA showed the same 11 repeat units in P97 R1 (repeat motif encoding AAKP[EV]), and 8 repeat units for H4 (QTTQKD). Regarding P146 R3, tandem repeats with 26, 27 and 28 units (S) were identified in samples from June 2013, whereas all typed samples from August 2015 showed 26 repeat units (Table 1).
Mycoplasma hyopneumoniae detection and typing using DNA extracted from nasal swabs and broncho-alveolar lavages (BAL) of sows and pigs at different ages and at slaughtera
| nPCR for M. hyopneumoniae detection (positive samples/analyzed samples) | MLVA loci | ||||
|---|---|---|---|---|---|
| P97 R1 | P146 R3 | H4 | |||
| Number of repeat units (number of samples) | Number of repeat units (number of samples) | Number of repeat units (number of samples) | |||
| June 2013 | Sows (nasal swabs) | 4/30 | None positive | 26 (1) | None positive |
| Pigs (nasal swabs) | 17/105 | 11(4) | 26 (8), 27 (1), 28 (1) | 8 (1) | |
| Slaughter (BAL) | 6/30 | 11(4) | 26 (6) | 8 (4) | |
| August 2015 | Pigs (nasal swabs) | 3/45 | 11(1) | None positive | 8 (1) |
| Slaughter (BAL) | 5/30 | 11(5) | 26 (5) | 8 (5) | |
A two-site, 1300-sow, commercial wean-to-finish farm was monitored looking for M. hyopneumoniae in cross-sectional studies carried out in June 2013 and in August 2015. In these studies, some nasal swabs and broncho-alveolar lavages of sows and pigs had rendered nPCR positive results. Typing was performed to assess the genetic diversity from some of these clinical specimens. The number repeat units of p97, p146 and H4 genes was determined by a nested-polymerase chain reactions as following: For P146 using as outer primers those described by Tamiozzo et al.8, and as inner primers those described by Mayor et al.6 Sequencing was performed using this last primer pair. For P97 and H4 using as outer primers those described by de Castro et al.2, and as inner primers those described by Vranckx et al.10 Sequencing was performed using this last primer pair.
Considering that all the samples from 2015 analyzed in the present study showed the same 3-loci profile as most samples taken in 2013, persistence of a M. hyopneumoniae strain over this period is highly suggested. The high discriminatory power reported by Dos Santos et al.3, for two of these loci (P97 R1 and P146 R3) gives support to that conclusion. Furthermore, the MLVA approach was reinforced in the present study by adding another polymorphic locus (H4).
Persistence of same MLVA type of M. hyopneumoniae over time is not an unexpected result due to the fact that it was a closed herd and the presence of only one distinct strain within the herds and low genetic diversity at herd-level has already been reported11. The access of another similar genetic strain through personnel, fomites or airborne transmission is highly improbable, because the presence of M. hyopneumoniae strain with 26 polyserine repeat motif of p146 gene had never been found in our country.
Apparently, the strain circulated among the pig population without any pressure of selection in the genome, or at least in the loci that were analyzed, in spite of vaccination and antibiotic treatment of the pigs. This may result in the persistence of the same genetic type. In this way a recent study has demonstrated the stability of P97 R1 and P146 R3 as in vivo as in vitro3.
Kuhnert and Overesch4 have shown, using MLST typing, the persistence of one M. hyopneumoniae strain in some farms, causing recurrent outbreaks, relating this fact to unsuccessful sanitation measures. In addition, it has been determined that M. hyopneumoniae can remain in pig organs even after antibiotic treatment5. More in-depth studies are necessary to determine the role of these MLVA loci in the development of mechanisms of persistence and evasion of the host immune system.
The VNTR PCR tests implemented in a nested format proved to be useful to type M. hyopneumoniae from clinical specimens without killing animals or performing invasive samplings. Considering that the 3-loci MLVA profile detected in the present study cannot be easily compared with results from other studies because of not all these three loci were analyzed or the number of repeat units present were not determined3,6,7; we consider that the use of a standardized nomenclature for MLVA loci and alleles of M. hyopneumoniae is highly needed to aid comparison of results from different studies.
To the best of our knowledge, this is the first report showing the persistence of a same genetic type of M. hyopneumoniae for such a long time period. We are aware that knowledge of M. hyopneumoniae persistence and genetic variability are essential to understand some epidemiological aspects of enzootic pneumonia and to develop and evaluate strategies for disease control.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appears in this article.
FundingThis study was financially supported in part by PICT 0442/2015 FONCyT-ANPCyT-MinCyT, República Argentina and PPI 2016–2018, 188/2016- UNRC. F. Rebaque was holder of an “Incentivo a las Vocaciones Científicas” scholarship during the development of this study (CIN-SPU-Ministerio de Educación), República Argentina.
Conflict of interestNone reported.
