









Tuberculosis remains a serious threat to human health as an infectious disease in Mexico. Data about the genotypes of circulating Mycobacterium tuberculosis isolates (MTB) in the State of Nuevo Leon, Mexico are scarce. We aimed to determine the genotypes of circulating MTB belonging to the Beijing lineage recovered from patients in the State of Nuevo Leon, Mexico. A total of 406 MTB isolates from this state were genotyped using the spoligotyping method and 18-locus MIRU-VNTR. Lineage classification and MTB transmission analysis were performed. Based on the spoligotyping analysis, we found 24 strains belonging to the Beijing genotype that were characterized phylogenetically. The MIRUs showed greater discriminatory power than the standard RFLP-IS6110 method; therefore, the greatest allelic diversity among the Beijing strains was observed with MIRU10, MIRU31, MIRU39, MRU40, and MIRU 26. MVLA analysis showed a profile variation between Beijing and non-Beijing strains. The minimum spanning tree (MST) showed that 79% (19) of the strains are related. All Beijing strains exhibited the deletion of region TbD1, which is a characteristic of modern strains. The application of spoligotyping and MIRU-VNTR-18 methods together proved to be more sensitive, discriminatory, and rapid than the standard method for the epidemiological analysis of Mycobacterium Beijing isolates. This study is one of the first to describe the genomic diversity of M. Beijing in the State of Nuevo Leon, Mexico.
La tuberculosis sigue siendo una grave amenaza para la salud humana como enfermedad infecciosa en México. Los datos sobre los genotipos de los aislados de Mycobacterium tuberculosis (MTB) circulantes en el estado mexicano de Nuevo León son escasos. Nuestro objetivo fue determinar los genotipos circulantes de MTB pertenecientes al linaje Beijing recuperados de pacientes en ese estado. Un total de 406 aislamientos de MTB del estado de Nuevo León, México, fueron genotipados con el método espoligotipado y MIRU-VNTR de 18 locus. Se realizaron clasificación de linaje y análisis de transmisión de MTB. Con base en el análisis de espoligotipado, encontramos 24 cepas pertenecientes al genotipo Beijing y se caracterizaron filogenéticamente. Los MIRU mostraron un mayor poder discriminatorio que el método estándar de RFLP-IS6110, mayor diversidad alélica entre las cepas de Beijing se observó con MIRU10, MIRU31, MIRU39, MRU40 y MIRU 26. El análisis de MVLA mostró una variación de perfil entre las cepas de Beijing y no Beijing. El árbol de expansión mínima (MST) mostró que el 79% (19) de las cepas están relacionadas. Todas las cepas de Beijing tenían la deleción de la región TbD1, característica de las cepas modernas. La aplicación de los métodos de espoligotipado y MIRU-VNTR-18 en conjunto resultó ser más sensible, discriminatoria y rápida que el método estándar para el análisis epidemiológico de aislamientos de Mycobacterium Beijing. Este estudio es uno de los primeros en describir la diversidad genómica de Mycobacterium Beijing en el Estado mejicano de Nuevo León.
Since Mycobacterium tuberculosis genotype Beijing was found as the causative agent of the drug-resistant tuberculosis outbreak in the 1990s, there was increased interest to know the origin, epidemiology, and specific characteristics of the Beijing strain, which appears to have higher virulence and greater spread ability. M. tuberculosis lineage 2, known as the Beijing lineage, is particularly abundant in Asian countries,27 and is considered a clonal population derived from a common ancestor, spreading radially from Beijing to other geographic areas.16 Several reports show that Beijing strains are now distributed worldwide, and are often associated with multidrug resistance.2,11 The higher resistance characteristic of these strains is possibly due to an increased rate of mutation.8,12
A minor sublineage, known as variant 0, belonging to a branch of the Beijing family, caused an outbreak in HIV+ patients in penitentiaries and hospitals at New York City in the 1990s and was designated as the W strain. Similarly to this case, there are several variants around the world (Bifani 2321, Kurepina 31).
Diverse animal models have shown that Beijing strains are more virulent, have a higher growth rate, produce more histopathological changes, and more deaths than other strains. They have special characteristics in their protein and lipid structures and in the way they interact with the immune system. Moreover, this group has been associated with specific polymorphisms in human genes related to the immune response suggesting a co-evolution with humans.19
However, the heterogeneity of these organisms in terms of drug-resistance, virulence (Dormans 137), and epidemiological characteristics suggests a genetic diversity leading to several sublineages.20 This diversity has been related to the fact that the dissemination of this genotype from its original focus radiated worldwide in several waves, mainly in the 19th century; and the increased drug resistance came as a result of the weakness of health systems, indiscriminate use of antibiotics, the emergence of the HIV/AIDS epidemic and an increase in the pathogen's population size.16
Based on the presence or absence of the IS6110 sequence in the NTF (noise transfer function) region, the Beijing strains are classified into modern or typical strains and ancestral or atypical strains.1 Among the modern strains are those containing one IS6110 sequence in the NTF region and the W strain with a double insertion, while the ancestral strains do not have these insertions.17 Large sequence polymorphism (LSPs) analysis identified differences among Beijing strains.25 The RD105 and RD207 regions are absent in all the Beijing family, whereas the RD142, RD150 and RD181 regions divide the family into 4 monophyletic groups.9,25 The RD181 deletion occurred early during the evolution of the lineage, while the RD150 and RD142 deletions have a low value for the phylogeny.6 The relationship between the members of the Beijing family, the emergence of drug resistance, the increased virulence, and their spreading ability justify the effort to characterize the isolates to promptly detect the presence of high-risk variants that could potentially affect individuals. In this study, spoligotyping and 18-locus MIRU-VNTR methods were used to identify different strains of Mycobacterium Beijing isolated from humans, with the aim to better analyze genetic diversity and determine their dominant types in the State of Nuevo Leon, Mexico.
Materials and methodsSamplingIn this study, 406 isolates of M. tuberculosis recovered from sputum samples of patients diagnosed with pulmonary tuberculosis were analyzed by using the spoligotyping technique.
Ethical approvalFor this study, all experiments were performed in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants and ethical approval was obtained from the Research Ethics Committee of the Faculty of Medicine and Psychology of the Autonomous University of Baja California (ref.: 2046).
All of them were isolated in the State of Nuevo Leon, Mexico from 2008 to 2015. Samples were processed using the Petroff method and seeded into Löwenstein–Jensen medium and MGIT liquid medium. All mycobacteria were characterized by conventional tests (niacin, catalase, and nitrates or immunochromatography) at the State Public Health Laboratory of Nuevo Leon, Mexico. Genomic DNA was obtained from those isolates identified as M. tuberculosis. Two small loopfuls of mycobacterial culture were suspended in 1ml of lysis solution (Promega) and heated at 80°C and then cells were lysed using a Savant Bio 101 FastPrep FP120 cell disruption system (Gemini Lab.Apeldoorn, The Netherlands) by applying five cycles of 45s at a speed setting 6 with 2-min rest periods on ice in between runs. Mycobacterial DNA was isolated using the Wizard SV Genomic DNA Purification System A2365 (Promega), following the manufacturer's instructions. The isolated DNA was stored in 50μl of sterile nuclease-free water at 4°C until its use. DNA quality was evaluated by gel electrophoresis and concentration was measured from 1μl of the resuspended samples on a Nanodrop equipment (Thermo Scientific, Massachusetts, United State).
SpoligotypingSpoligotyping of all isolates was performed by amplifying the DR region from a 20ng/50μl dilution of extracted DNA, in nuclease-free water, using the standard method described by Kamerbeek et al.14 PCR amplicons were subjected to inverse hybridization analysis (Supplementary data (Table S1)), using a commercial membrane (Mapmygenome, India) that included spacer probes 1–43.14 Hybridation profile was revealed by chimioluminiscence with the ECL system (Enhanced Chemiluminescent detection) (Amersham Biosciences, Buckinghamshire, United Kingdom). M. tuberculosis and Mycobacterium bovis BCG were the controls for each experiment. The obtained profiles were compared with the spoligotyping international database SpolDB4.0. The strains selected for this work were those exhibiting a spoligotyping profile belonging to the Beijing family and having the RD181 deletion.
Drug susceptibility testTests were performed in a Bactec 960 equipment, following the manufacturer's instructions. Resistant and sensitive strains for each drug were included in each test as controls.
Multiple loci VNTR analysis (MLVA)Eighteen chromosomal regions containing tandem repeats were amplified by PCR.24,23 Primers for the 18 MIRU-VNTR loci were used, including twelve standard primers and six additional primers for the characterization of the Beijing genotype (Supplementary data (Table S2)). The amplicons were analyzed in 2% agarose gel electrophoresis. The number of repeats according to their size against a MW marker (100pb ladder, Promega, lugar) were obtained for each locus and then a code of 18 numbers was generated and analyzed using Bionumerics v7 software (Applied Maths NV, Sint-Martens-Latem, Bélgica) and a minimum spanning tree (MST) was constructed to identify the main lineages.
Single nucleotide polymorphism (SNPs) assaykatG and gyrA genes were analyzed by sequencing PCR fragments that were amplified from these specific regions using the primers shown in Supplementary data (Table S3). Sequences were aligned with the software BioEdit to identify SNPs described by Sreevatsan (1977).
Analysis of pks15/1 presenceA 144-pb segment of gene pks15/1 amplified by PCR was sequenced using the Sanger method (1977) and the resulting sequence was aligned using the Mega software (available online https://www.megasoftware.net) and the Muscle algorithm to identify the presence of a specific 7-bp sequence characteristic of the East Asian lineage.
Deletion of the TbD1 geneA nested PCR to amplify a 2000-bp region of gene TbD1 was conducted according to Brosch et al. (2002)3 and the resulting products were visualized on a 2% agarose gel (Supplementary data (Table S4)).
Large sequence polymorphism (LSPs) analysisThe presence of large sequence polymorphisms in regions RD105, 181, 207, 150 and 142 was determined by Real Time PCR on an AB 7500 equipment (Applied Biosystem).
Analysis of the noise transfer function (NTF) regionThe analysis of the NTF region was performed by PCR amplification, using the primers described by Plikaytis (1994) and modified by Wada (2009). The resulting PCR products were visualized on a 2% agarose gel (Supplementary data (Tables S5 and S6)).
ResultsBased on the spoligotyping analysis, we found fourteen strains belonging to the Beijing genotype, which were characterized phylogenetically. A group of 10 strains isolated from patients attended in the State of Baja California, and previously identified as Beijing, were included in this analysis to compare the profiles (Table 1). The percentages of the most frequently found genotypes were as follows: 38.9% belonged to the T lineage, 11.3% to the LAM lineage, 25.2% to the X lineage, 11% to the H lineage, 1% to the EAI2-Manila family, 2.9% to the S lineage and 0.6% to the U lineage. The remaining he strains belonged to 15 genotypes that were not identified in the database.
Spoligotypes shared by Mycobacterium tuberculosis strains evaluated in this study.
| Strain | Spoligotyping profile | Octal code | SIT | Clade |
|---|---|---|---|---|
| 349 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 362 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 376 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 394 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 524 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 545 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 110 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 113 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 115 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| H37Rv | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 777777477760771 | 451 | CONTROL |
| 2 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 19 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 22 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 25 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 27 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 45 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 53 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 59 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 66 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 70 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 593 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 710 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 130 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 123 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
| 129 | □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ | 000000000003771 | 1 | BEIJING |
SIT: poligotype international types; □: absence of spacer; ■: presence of spacer; O: orphan.
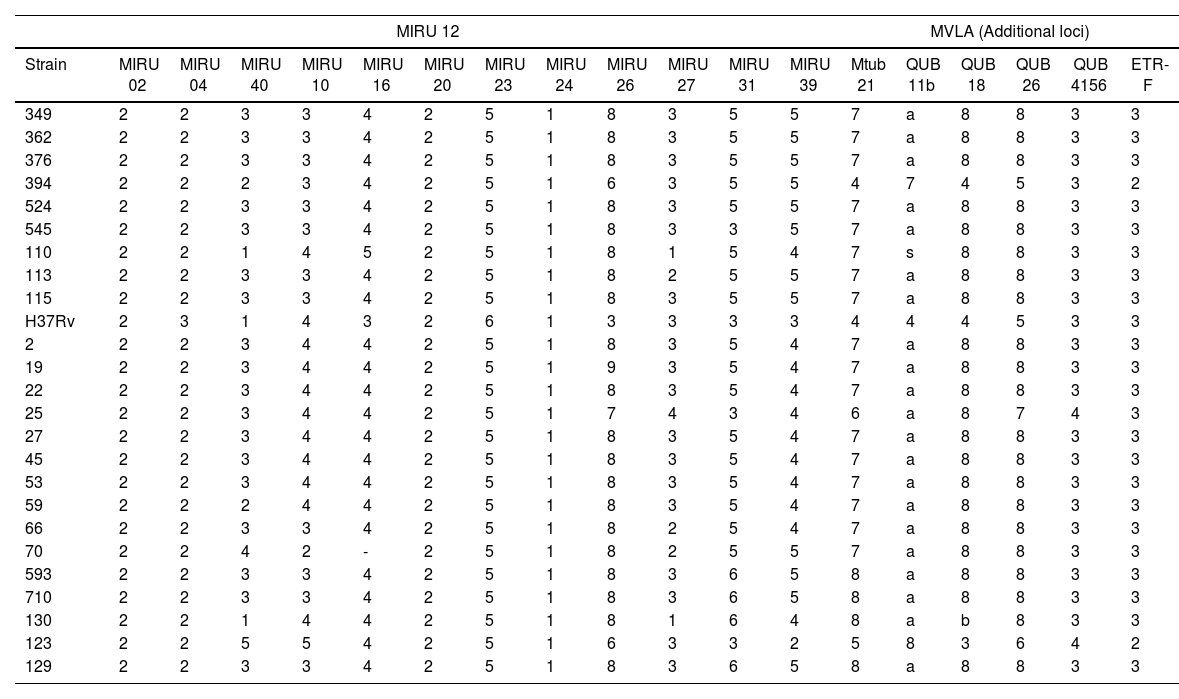
The MVLA analysis showed profile variations between Beijing and non-Beijing strains. Loci Mtub21, QUB11b y QUB18 revealed differences in Beijing and non-Beijing (123, 391 and H37Rv) strains. Non-Beijing strains had fewer repeats. MIRUs exhibited a higher discriminatory power and therefore the biggest allelic diversity among Beijing strains was observed in MIRU10, MIRU31, MIRU39, MRU40, and MIRU 26. Those lacking discriminatory power were MIRU 2, MIRU 10, MIRU 20, MIRU 23 Y MIRU 24 (Table 2).
Allele diversity of the 18-locus MIRU-VNTR in 24 Mexican Mycobacterium tuberculosis Beijing strains.
| MIRU 12 | MVLA (Additional loci) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain | MIRU 02 | MIRU 04 | MIRU 40 | MIRU 10 | MIRU 16 | MIRU 20 | MIRU 23 | MIRU 24 | MIRU 26 | MIRU 27 | MIRU 31 | MIRU 39 | Mtub 21 | QUB 11b | QUB 18 | QUB 26 | QUB 4156 | ETR-F |
| 349 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 5 | 7 | a | 8 | 8 | 3 | 3 |
| 362 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 5 | 7 | a | 8 | 8 | 3 | 3 |
| 376 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 5 | 7 | a | 8 | 8 | 3 | 3 |
| 394 | 2 | 2 | 2 | 3 | 4 | 2 | 5 | 1 | 6 | 3 | 5 | 5 | 4 | 7 | 4 | 5 | 3 | 2 |
| 524 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 5 | 7 | a | 8 | 8 | 3 | 3 |
| 545 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 3 | 5 | 7 | a | 8 | 8 | 3 | 3 |
| 110 | 2 | 2 | 1 | 4 | 5 | 2 | 5 | 1 | 8 | 1 | 5 | 4 | 7 | s | 8 | 8 | 3 | 3 |
| 113 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 2 | 5 | 5 | 7 | a | 8 | 8 | 3 | 3 |
| 115 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 5 | 7 | a | 8 | 8 | 3 | 3 |
| H37Rv | 2 | 3 | 1 | 4 | 3 | 2 | 6 | 1 | 3 | 3 | 3 | 3 | 4 | 4 | 4 | 5 | 3 | 3 |
| 2 | 2 | 2 | 3 | 4 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 4 | 7 | a | 8 | 8 | 3 | 3 |
| 19 | 2 | 2 | 3 | 4 | 4 | 2 | 5 | 1 | 9 | 3 | 5 | 4 | 7 | a | 8 | 8 | 3 | 3 |
| 22 | 2 | 2 | 3 | 4 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 4 | 7 | a | 8 | 8 | 3 | 3 |
| 25 | 2 | 2 | 3 | 4 | 4 | 2 | 5 | 1 | 7 | 4 | 3 | 4 | 6 | a | 8 | 7 | 4 | 3 |
| 27 | 2 | 2 | 3 | 4 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 4 | 7 | a | 8 | 8 | 3 | 3 |
| 45 | 2 | 2 | 3 | 4 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 4 | 7 | a | 8 | 8 | 3 | 3 |
| 53 | 2 | 2 | 3 | 4 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 4 | 7 | a | 8 | 8 | 3 | 3 |
| 59 | 2 | 2 | 2 | 4 | 4 | 2 | 5 | 1 | 8 | 3 | 5 | 4 | 7 | a | 8 | 8 | 3 | 3 |
| 66 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 2 | 5 | 4 | 7 | a | 8 | 8 | 3 | 3 |
| 70 | 2 | 2 | 4 | 2 | - | 2 | 5 | 1 | 8 | 2 | 5 | 5 | 7 | a | 8 | 8 | 3 | 3 |
| 593 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 6 | 5 | 8 | a | 8 | 8 | 3 | 3 |
| 710 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 6 | 5 | 8 | a | 8 | 8 | 3 | 3 |
| 130 | 2 | 2 | 1 | 4 | 4 | 2 | 5 | 1 | 8 | 1 | 6 | 4 | 8 | a | b | 8 | 3 | 3 |
| 123 | 2 | 2 | 5 | 5 | 4 | 2 | 5 | 1 | 6 | 3 | 3 | 2 | 5 | 8 | 3 | 6 | 4 | 2 |
| 129 | 2 | 2 | 3 | 3 | 4 | 2 | 5 | 1 | 8 | 3 | 6 | 5 | 8 | a | 8 | 8 | 3 | 3 |
MIRU-VNTR: mycobacterial interspersed repetitive unit variable number tandem repeat; MLVA: multiple loci VNTR analysis.
The minimum spanning tree (MST) showed that 79% (19) of the strains are related. In Figure 1, strains belonging to the same cluster are connected by a black line and are part of a clonal complex. The first cluster comprises strains 593, 710 y 129; a second cluster includes strains 2, 22, 27, 45 and 53; and a third cluster strains 349, 362, 376, 115 and 524. Twenty-one percent (five) are unrelated strains (25, 394, 110, 130 and 123) and are connected by a dotted line. All Beijing strains had the deletion of region TbD1, characteristic of modern strains.3
 showed than 79% (19) of the strains are related.")
Figure 2 shows the alignment of amplified pks15/1 gene sequences. Strains belonging to lineage 2, including the Beijing genotype, maintain the pks15/1 gene intact; while those showing a 7-bp deletion belong to other lineages, such as strains 394, 123 and H37Rv. Interestingly, two strains identified as Beijing by spoligotyping did not show an intact pks15/1 gene. The analysis of the NTF region with respect to the number of IS6110 insertions classified the Beijing strains as modern, except for strains 123 and 394, which were considered pseudo-Beijing based on this result and other additional observations. Most of the strains exhhibited two insertions (Table 3).

Amplification fragments of the insertion of IS6110 sequence into the NTF region.
| Plikaytis | WADA 2009 | |||||||
|---|---|---|---|---|---|---|---|---|
| ID | 523PB | 223PB | 175PB | IS6110 copies | 302PB | 280PB | 1500PB | Classification |
| H37Rv | □ □ | 0 | □ □ | □ □ | N/A | |||
| 394 | □ □ | 0 | □ □ | □ □ | N/A (pseudo-Beijing) | |||
| 123 | □ □ | 0 | □ □ | □ □ | N/A (pseudo-Beijing) | |||
| 110 | □ □ | □ □ | 1 | □ □ | MODERN | |||
| 22 | □ □ | □ □ | 1 | □ □ | MODERN | |||
| 25 | □ □ | □ □ | 1 | □ □ | MODERN | |||
| 593 | □ □ | □ □ | 1 | □ □ | MODERN | |||
| 130 | □ □ | □ □ | 1 | □ □ | MODERN | |||
| 349 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 362 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 376 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 524 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 545 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 113 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 115 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 2 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 19 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 27 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 45 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 53 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 59 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 66 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 710 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||
| 129 | □ □ | □ □ | □ □ | 2 | □ □ | MODERN | ||

All the isolates identified as Beijing strains by spoligotyping were subjected to LSPs analysis. The RD 105, 207 and 181 regions were deleted in all of them (Fig. 3). Based on the subsequent deletion of RD 150 and 142, the strains were grouped into three classes: (1) strains with both RD 150 and 142 deleted (1 strain); (2) strains with RD 150 deleted and RD 142 present (13 strains); (3) strains with both RD 150 and 142 present. Phylogenetically unrelated strains constitute 8% of the total (strains 394 and 123). These results are shown in Figure 3.
 and lineage 4 Euro-American (in red). Lineage 2 is represented by a continuous blue line emerging from the root and with the deletions RD105, RD207 and RD181; this line divides into 3 clades, the first one has the RDs 150 and 142, the second clade lacks RD 150 but has RD 142, while the third clade lacks both RDs 150 and 142. Lineage 4, represented by a red line emerging from the root, has RDs 105, 207, 181, 150 and 142.")
The strains analyzed in this study came from a common ancestor of M. tuberculosis, which branched into lineage 2 from Eastern Asia (in blue) and lineage 4 Euro-American (in red). Lineage 2 is represented by a continuous blue line emerging from the root and with the deletions RD105, RD207 and RD181; this line divides into 3 clades, the first one has the RDs 150 and 142, the second clade lacks RD 150 but has RD 142, while the third clade lacks both RDs 150 and 142. Lineage 4, represented by a red line emerging from the root, has RDs 105, 207, 181, 150 and 142.
The M. tuberculosis Beijing genotype is the predominant strain in many Asian countries. Due to its rapid spread worldwide, it is gaining significant attention. Moreover, its connection to drug resistance, increased virulence and faster transmission rate highlights the epidemiological relevance of identifying this strain. The strain was first described in the 1990s as strain “W”, being the causative agent of tuberculosis outbreaks in New York. The analysis revealed that the strain was a variant originating in East Asia. Genetic profile studies using specific markers have shown a wide variability within the Beijing family, resulting in different profiles of drug resistance and virulence.18
The multidrug resistant M. tuberculosis W strains that were isolated during the outbreaks in New York carried two IS6110 sequence insertions in the NTF region, and are considered modern strains, unlike the common strains in Asia carrying only one IS6110 insertion and considered ancestral strains.17,28 Among our isolates, we found five strains with a single IS6110 insertion and 16 strains with two IS6110 insertions. Probably, the strains with two insertions were derived from those with one insertion. Strain variants may emerge constantly, but only some of them exhibit greater drug resistance, improved virulence, and transmission capabilities, making them more successful to spread within the population. Although Beijing strains are associated with drug resistance, our results show that strains isolated in the State of Nuevo Leon are susceptible to first-line drugs, with only one strain displaying resistance to isoniazid.
The absence of specific regions in mycobacteria is the result of unique and irreversible events producing biological markers that allow to build robust phylogenies.10,13,25 Beijing strains from lineage 2 lack RD 105, which identifies them as coming from East Asia. This lineage also lacks RD207, which includes spacers 1 to 34, thereby giving them the distinctive spoligotyping profile. Additionally, some variants have lost the RD181 region, and are considered modern Beijing strains; among them, some have also lost RD142 and RD150.4 The mycobacteria isolated in this work are considered modern strains as they have two IS6110 insertions in the NTF region and lack the TbD1 region. Two strains of this group had the RD181 and RD105 regions, but lack the RD207 region.
Ninety-two percent (22) of the isolates belong to phylogenetic group 1; four percent (1) to group 2 and four percent (1) to group 3 according to the Sreevatsan classification.21 We originally expected that all strains were classified in group 1 (East Asia), but the analysis showed that strain 123 (group 2) and 394 (group 3) have a 7-bp deletion in the pks15/1 gene, which the Beijing strains do not have. The pks15/1 gene is involved in the biosynthesis of phenolic glycolipids (PGLs) and is polymorphic in members of M. tuberculosis complex.5 While Beijing strains have the pks15/1 gene intact, strain H37Rv has the 7-pb deletion and belongs to group 3.3,5,15,21
All these data altogether showed that the group of strains isolated in this work belongs to the Beijing family, although they lack all the typical characteristics of the Beijing genotype and therefore have a different phylogenetic origin. Loss of the RD207 region is due to the insertion of the IS6110 sequence into the RD207 region that may cause the partial or complete loss of RD207.7,25 Different authors found that strains with spolygotyping lacking spacers 1–34 lost a bigger or smaller region than the equivalent to RD207 (7399bp) and this happens in strains not belonging to group 1 of the Sreevatsan classification,21 which are known as pseudo-Beijing strains. We observed the same in this study and the loss of spacers 1–34 may have been produced by convergent evolution as suggested.7 The latter study included pseudo-Beijing strains within lineage 3; however, we found that our strains belong to lineage 4, suggesting a higher prevalence of group 4 (Euro-American) in this geographic region.
In order to understand the phylogenetic relationship among the isolates, the number of clusters and the unique genotypes a minimum spanning tree (MST) was constructed using the Bionumerics v7 software (Applied Maths NV, Sint-Martens-Latem, Belgium). According to this analysis, it is highly likely that the infection of patients with unique strains was the result of sporadic contact, reactivation of that sporadic contact or imported cases. Moreover, infections caused by strains grouped into clusters exhibit a clonal relationship, although we could not establish a direct transmission, as they were collected in different years. Probably, this genotype has existed in the region for several decades, becoming an endemic genotype isolated sporadically. Additionally, most isolates are sensitive to first-line drugs.26,22
The MIRU-VNTR analysis showed that the MIRUs with higher allelic diversity and therefore better discriminatory power to distinguish Beijing strain sublineages were MIRU10, MIRU31, MIRU39, MRU40, and MIRU 26. The variability found with these markers suggests that the isolates did not have the same origin. The dendrogram based on the 18 MLVAs reveals that strains 123 and 394 do not have any relationship with Beijing strains and that the isolates from the State of Nuevo Leon and those reported from Baja California belong to two different populations. Strains 110 and 130 did not belong to any of these groups and, therefore, they might be imported or reactivation cases. The State of Baja California has a high commercial activity with Asian countries. Tijuana is one of the main seaports in Mexico and during the past year, thousands of Central American immigrants have arrived in the city. Mexicali, located 180km away from Tijuana, houses the largest Chinese community in Mexico. Trade and human immigration from these areas might have facilitated the introduction of the Asian lineage and its later dissemination to the rest of the country, which could have resulted in the generation of sublineages as was observed in this study. Records from the international spoligotyping database up to 2006 showed than Beijing and Beijing-like strains represent around 50% of the strains in Eastern Asia and 13% of the isolates worldwide.19 The percentage of the Beijing genotype in previous studies had been close to 1.3%. However, among the 406 strains analyzed in this study, the percentage was 3.4%, implying a dissemination of the genotype, yet this prevalence has not manifested itself as an outbreak.
FundingThe authors acknowledge project 568475 of the Frontier Sciences Fund-CONACYT for supporting this research, and University of Monterrey for financing the publication fees.
Authors’ contributionsPBM and FGS – funding acquisition; NCB, JCG, HAR, MBM, AMV, VVM – methodology; NCB, PBM and VVM – data curation; NCB, PBM and VVM – formal analysis; JCG, HAR, MBM and AMV – supervision; NCB, PBM, FGS, HAR and VVM – manuscript writing; review and editing – all authors.
Conflict of interestsThe author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors would like to express their gratitude to the personnel of Health of the State of Nuevo Leon, Mexico for support and collaboration in this study.
The followings are the supplementary data to this article:
