



Bacterial richness in maritime Antarctica has been poorly described to date. Phylogenetic affiliation of seawater free-living microbial assemblages was studied from three locations near the Argentinean Jubany Station during two Antarctic summers. Sixty 16S RNA cloned sequences were phylogenetically affiliated to Alphaproteobacteria (30/60 clones), Gammaproteobacteria(19/60 clones), Betaproteobacteria and Cytophaga–Flavobacteriia–Bacteroides (CFB), which were (2/60) and (3/60) respectively. Furthermore, six out of 60 clones could not be classified. Both, Alphaproteobacteria and Gammaproteobacteria, showed several endemic and previously undescribed sequences. Moreover, the absence of Cyanobacteria sequences in our samples is remarkable. In conclusion, we are reporting a rich sequence assemblage composed of widely divergent isolates among themselves and distant from the most closely related sequences currently deposited in data banks.
La riqueza bacteriana de la Antártida marítima ha sido pobremente descripta hasta la actualidad. En este trabajo se estudió la filogenia de un grupo de colecciones bacterianas planctónicas obtenidas de agua de mar de tres localizaciones cercanas a la base científica argentina antártica Jubany (actualmente Carlini). Sesenta secuencias clonadas del ARN 16S fueron agrupadas filogenéticamente en las clases Alfaproteobacteria (30/60 clones), Gammaproteobacteria (19/60 clones), Betaproteobacteria (2/60) y Cytophaga–Flavobacteriia–Bacteroides [CFB (3 / 60)]. Por otro lado, seis de las sesenta secuencias (6/60) no pudieron ser clasificadas en ningún grupo conocido. Tanto las Alfaproteobacteria como las Gammaproteobacteria mostraron secuencias no descriptas con anterioridad y eventualmente endémicas. También es remarcable la ausencia de Cyanobacteria en esta biblioteca. En conclusión, estamos publicando aquí una rica colección de secuencias bacterianas de origen marino compuesta por una mayoría de secuencias ampliamente divergentes entre sí y también distantes de aquellas más intimamente relacionadas dentro de las depositadas hasta el presente en los bancos de datos.
Because of the vast extension of the oceans, all biochemical processes occurring in marine ecosystems have a significant effect on global biogeochemical cycles of carbon, sulphur, nitrogen and other elements. These cycles are driven by marine microbes that have been subjected to a strong selective pressure over long evolutionary timespans27,30. In this context, the unraveling of microbial community structure and functions in the world's oceans is one of the most fascinating chapters in ecology.
As extreme environments are explored, the richness of microbial communities is increasingly evident. Since the knowledge of microbial diversity is crucial to the maintenance and conservation of global genetic resources, studies dealing with the composition and structure of microbial communities inhabiting such extreme environments represent a key step in the understanding of these previously poorly analysed environments. This fact highlights the relevance of the studies on bacterial diversity in Antartica, some of whose biotopes represent the coldest sites in the biosphere8.
As biodiversity can be affected by environmental disturbances from both natural and anthropogenic origin, regulation measures are required to promote and conduct studies on microbial Antarctic diversity leading to a rational record of their components. The present interest in this problem is reflected by the text of Recommendation No. 20 of the Antarctic Treaty Meeting of Experts (ATME) on Climate Change arising from the 13th Meeting of the Committee for Environmental Protection (CEP) held in Punta del Este, Uruguay in May 2010.
Free-living marine bacterioplankton communities in near-surface layers seem to be dominated by phyla Alphaproteobacteria and Gammaproteobacteria and members of classes Flavobacteriia and Sphingobacteriia of the Bacteroidetes phylum15,22. This dominance has also been reported for Antarctic ice, marine waters and sediments3–5.
Contrary to the deep ocean, polar marine environments undergo extreme temporal variations in sea ice cover, light levels and day length. These factors greatly influence the biology of these polar environments and result in strong biomass production in spring and summer followed by a long winter of very low rates of primary production21. Furthermore, polar environments have been shown to be highly sensitive to global warming, exhibiting significant effects on the extent and thickness of sea ice in response to minimal changes in the average temperature values. As our knowledge of the dynamics and diversity of Antarctic marine bacterioplankton is still in its infancy.
Antarctic SSU rRNA gene phylotypes frequently show a high degree of similarity with other polar sequences. In other cases, some species such as Polaribacter irgensii and also uncultivated members of Gammaproteobacteria (Ant4D3 and Ant10A4) seem to proliferate only in Antarctic waters, further and deeper studies being necesary to address diversity on a larger scale18.
In this study, phylogenetic affiliation of seawater free-living microbial assemblages from three sites in Potter Cove (King George Island), which were sampled during two consecutive Antarctic summers (January–March 2008 and 2009), were studied in order to describe the richness, in time and space, of the dominant bacterioplankton components inhabiting this maritime Antarctic area whose coastal zone is considered a Specially Protected Area (ASPA) 132 by the Scientific Committee on Antarctic Research (SCAR).
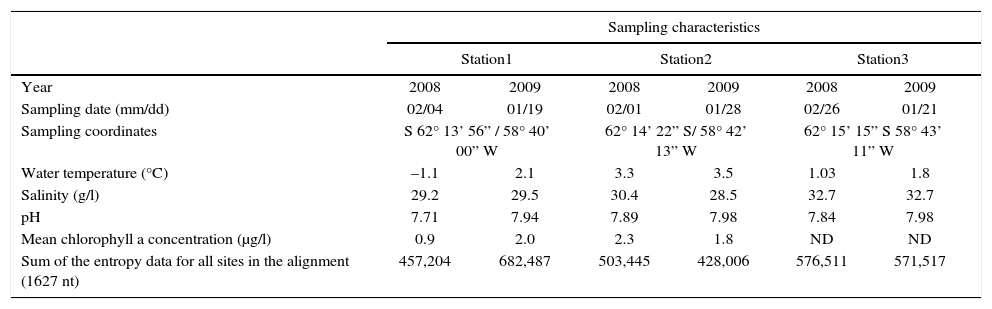
Materials and methodsSampling sitesSurface sea water (0-0.3m depth) was obtained from three different locations close to Jubany Station, on the coast of Potter Cove, King George Island (Isla 25 de Mayo), South Shetland Islands. The following data associated with each sample were recorded and are shown in Table 1: latitude and longitude, temperature, chlorophyll concentration, salinity, pH and sampling date.
Description of sampling sites and datesa
| Sampling characteristics | ||||||
|---|---|---|---|---|---|---|
| Station1 | Station2 | Station3 | ||||
| Year | 2008 | 2009 | 2008 | 2009 | 2008 | 2009 |
| Sampling date (mm/dd) | 02/04 | 01/19 | 02/01 | 01/28 | 02/26 | 01/21 |
| Sampling coordinates | S 62° 13’ 56” / 58° 40’ 00” W | 62° 14’ 22” S/ 58° 42’ 13” W | 62° 15’ 15” S 58° 43’ 11” W | |||
| Water temperature (°C) | –1.1 | 2.1 | 3.3 | 3.5 | 1.03 | 1.8 |
| Salinity (g/l) | 29.2 | 29.5 | 30.4 | 28.5 | 32.7 | 32.7 |
| pH | 7.71 | 7.94 | 7.89 | 7.98 | 7.84 | 7.98 |
| Mean chlorophyll a concentration (μg/l) | 0.9 | 2.0 | 2.3 | 1.8 | ND | ND |
| Sum of the entropy data for all sites in the alignment (1627 nt) | 457,204 | 682,487 | 503,445 | 428,006 | 576,511 | 571,517 |
Surface water samples (35 litres) were aseptically collected on surface and serially filtered onto 1,5μm and 0.2-μm-pore-size filters (140mm diameter; MSI, USA fiber glass and cellulose acetate respectivelly). Total DNA was extracted from a small piece of filters (10mm×30mm) using the phenol–chloroform protocol as described earlier16.
16S rRNA gene clone library construction16S rRNA gene from total DNA obtained from each location was amplified by PCR (denaturation step for 3min at 94°C; 40 cycles consisting of 60 s at 94°C, 60 s at 55°C and 120 s at 72°C; followed by 10min at 72°C) using Go Taq Polymerase and universal primers E8F (AGAGTTTGATCCTGGCTCAG) and E1541R (AAGGAGGTGATCCANCCRCA)1. PCR products (50 ng) were ligated into the pGEM-T Easy Vector (Promega) and used for transformation of competent E. coli DH5α cells. The transformed cells were plated on Luria-Bertani (LB) plates containing ampicillin (100μg/ml), 1.80μg of X-Gal (5-bromo-4-chloro-3-indolyl- beta-D-galactopyranoside)/ml–1, and 0.5mM IPTG (Isopropyl β-D-1-thiogalactopyranoside) as recommended by the manufacturer and incubated overnight at 37°C. Twenty four recombinant clones for each location and sampling time were picked and plasmid minipreps were made. For the selection of clones for further sequencing the following criteria were considered: a) insert length, b) presence or absence of EcoRI digestion sites and c) when present, the position of EcoRI cut sites. Finally, ten clones were sequenced for each time and location. In brief, sequenced clones were not selected at random.
RDP Naïve Bayesian rRNA Classifier Version 2.0, July 2007Hierarchical taxa were based on a naïve Bayesian rRNA classifier28. A 95% confidence threshold was used for isolate affiliation.
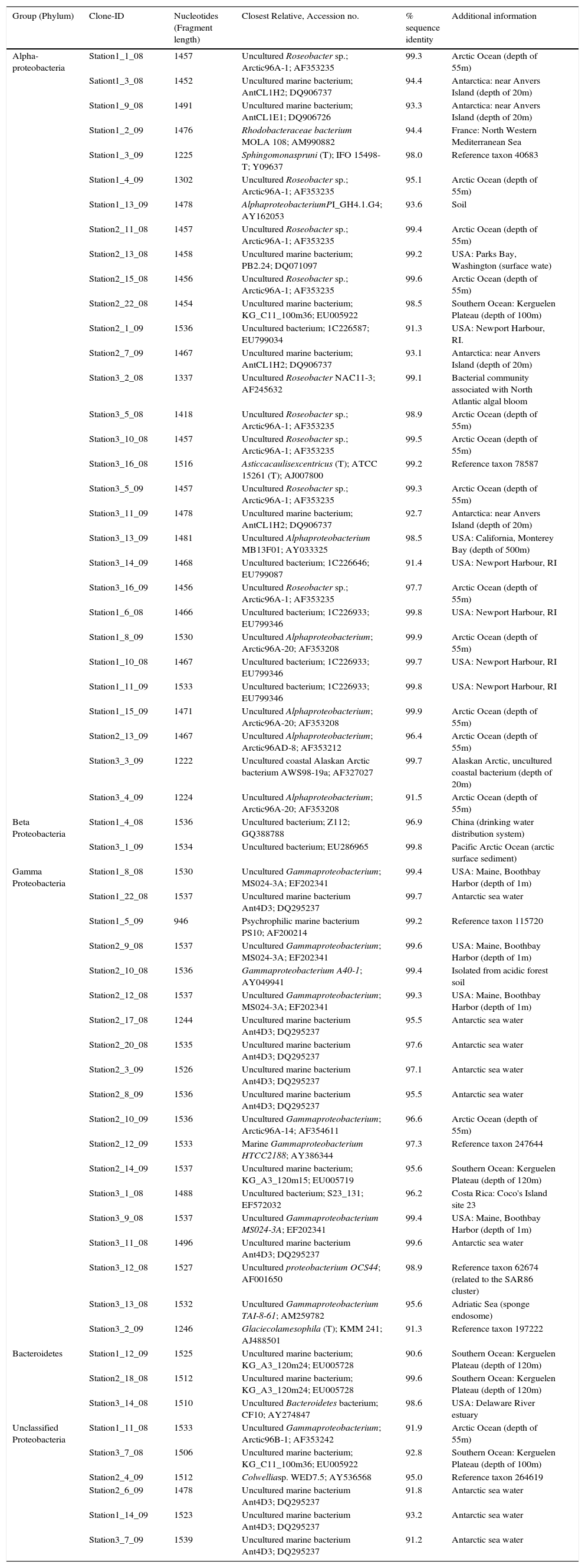
Identity matrix calculation and phylogenetic analysisIn this study, 60 clones were sequenced by using plasmid primer SP6 and also plasmid primer T7 to obtain nearly complete sequences of the 16S rRNA gene (length 1222-1539 nucleotides, Table 2). Primer sequences were not included in phylogenetic analyses. Sequences were checked for chimeras by generating phylogenetic trees with different regions of the sequence. Sequences were aligned and assigned to major groups (Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria) by using the RDP Database Project II Release 10.19 and molecular phylogeny in parallel.
Phylogenetic affiliation (phylum) and clone identification.
| Group (Phylum) | Clone-ID | Nucleotides (Fragment length) | Closest Relative, Accession no. | % sequence identity | Additional information |
|---|---|---|---|---|---|
| Alpha-proteobacteria | Station1_1_08 | 1457 | Uncultured Roseobacter sp.; Arctic96A-1; AF353235 | 99.3 | Arctic Ocean (depth of 55m) |
| Sationt1_3_08 | 1452 | Uncultured marine bacterium; AntCL1H2; DQ906737 | 94.4 | Antarctica: near Anvers Island (depth of 20m) | |
| Station1_9_08 | 1491 | Uncultured marine bacterium; AntCL1E1; DQ906726 | 93.3 | Antarctica: near Anvers Island (depth of 20m) | |
| Station1_2_09 | 1476 | Rhodobacteraceae bacterium MOLA 108; AM990882 | 94.4 | France: North Western Mediterranean Sea | |
| Station1_3_09 | 1225 | Sphingomonaspruni (T); IFO 15498-T; Y09637 | 98.0 | Reference taxon 40683 | |
| Station1_4_09 | 1302 | Uncultured Roseobacter sp.; Arctic96A-1; AF353235 | 95.1 | Arctic Ocean (depth of 55m) | |
| Station1_13_09 | 1478 | AlphaproteobacteriumPI_GH4.1.G4; AY162053 | 93.6 | Soil | |
| Station2_11_08 | 1457 | Uncultured Roseobacter sp.; Arctic96A-1; AF353235 | 99.4 | Arctic Ocean (depth of 55m) | |
| Station2_13_08 | 1458 | Uncultured marine bacterium; PB2.24; DQ071097 | 99.2 | USA: Parks Bay, Washington (surface wate) | |
| Station2_15_08 | 1456 | Uncultured Roseobacter sp.; Arctic96A-1; AF353235 | 99.6 | Arctic Ocean (depth of 55m) | |
| Station2_22_08 | 1454 | Uncultured marine bacterium; KG_C11_100m36; EU005922 | 98.5 | Southern Ocean: Kerguelen Plateau (depth of 100m) | |
| Station2_1_09 | 1536 | Uncultured bacterium; 1C226587; EU799034 | 91.3 | USA: Newport Harbour, RI. | |
| Station2_7_09 | 1467 | Uncultured marine bacterium; AntCL1H2; DQ906737 | 93.1 | Antarctica: near Anvers Island (depth of 20m) | |
| Station3_2_08 | 1337 | Uncultured Roseobacter NAC11-3; AF245632 | 99.1 | Bacterial community associated with North Atlantic algal bloom | |
| Station3_5_08 | 1418 | Uncultured Roseobacter sp.; Arctic96A-1; AF353235 | 98.9 | Arctic Ocean (depth of 55m) | |
| Station3_10_08 | 1457 | Uncultured Roseobacter sp.; Arctic96A-1; AF353235 | 99.5 | Arctic Ocean (depth of 55m) | |
| Station3_16_08 | 1516 | Asticcacaulisexcentricus (T); ATCC 15261 (T); AJ007800 | 99.2 | Reference taxon 78587 | |
| Station3_5_09 | 1457 | Uncultured Roseobacter sp.; Arctic96A-1; AF353235 | 99.3 | Arctic Ocean (depth of 55m) | |
| Station3_11_09 | 1478 | Uncultured marine bacterium; AntCL1H2; DQ906737 | 92.7 | Antarctica: near Anvers Island (depth of 20m) | |
| Station3_13_09 | 1481 | Uncultured Alphaproteobacterium MB13F01; AY033325 | 98.5 | USA: California, Monterey Bay (depth of 500m) | |
| Station3_14_09 | 1468 | Uncultured bacterium; 1C226646; EU799087 | 91.4 | USA: Newport Harbour, RI | |
| Station3_16_09 | 1456 | Uncultured Roseobacter sp.; Arctic96A-1; AF353235 | 97.7 | Arctic Ocean (depth of 55m) | |
| Station1_6_08 | 1466 | Uncultured bacterium; 1C226933; EU799346 | 99.8 | USA: Newport Harbour, RI | |
| Station1_8_09 | 1530 | Uncultured Alphaproteobacterium; Arctic96A-20; AF353208 | 99.9 | Arctic Ocean (depth of 55m) | |
| Station1_10_08 | 1467 | Uncultured bacterium; 1C226933; EU799346 | 99.7 | USA: Newport Harbour, RI | |
| Station1_11_09 | 1533 | Uncultured bacterium; 1C226933; EU799346 | 99.8 | USA: Newport Harbour, RI | |
| Station1_15_09 | 1471 | Uncultured Alphaproteobacterium; Arctic96A-20; AF353208 | 99.9 | Arctic Ocean (depth of 55m) | |
| Station2_13_09 | 1467 | Uncultured Alphaproteobacterium; Arctic96AD-8; AF353212 | 96.4 | Arctic Ocean (depth of 55m) | |
| Station3_3_09 | 1222 | Uncultured coastal Alaskan Arctic bacterium AWS98-19a; AF327027 | 99.7 | Alaskan Arctic, uncultured coastal bacterium (depth of 20m) | |
| Station3_4_09 | 1224 | Uncultured Alphaproteobacterium; Arctic96A-20; AF353208 | 91.5 | Arctic Ocean (depth of 55m) | |
| Beta Proteobacteria | Station1_4_08 | 1536 | Uncultured bacterium; Z112; GQ388788 | 96.9 | China (drinking water distribution system) |
| Station3_1_09 | 1534 | Uncultured bacterium; EU286965 | 99.8 | Pacific Arctic Ocean (arctic surface sediment) | |
| Gamma Proteobacteria | Station1_8_08 | 1530 | Uncultured Gammaproteobacterium; MS024-3A; EF202341 | 99.4 | USA: Maine, Boothbay Harbor (depth of 1m) |
| Station1_22_08 | 1537 | Uncultured marine bacterium Ant4D3; DQ295237 | 99.7 | Antarctic sea water | |
| Station1_5_09 | 946 | Psychrophilic marine bacterium PS10; AF200214 | 99.2 | Reference taxon 115720 | |
| Station2_9_08 | 1537 | Uncultured Gammaproteobacterium; MS024-3A; EF202341 | 99.6 | USA: Maine, Boothbay Harbor (depth of 1m) | |
| Station2_10_08 | 1536 | Gammaproteobacterium A40-1; AY049941 | 99.4 | Isolated from acidic forest soil | |
| Station2_12_08 | 1537 | Uncultured Gammaproteobacterium; MS024-3A; EF202341 | 99.3 | USA: Maine, Boothbay Harbor (depth of 1m) | |
| Station2_17_08 | 1244 | Uncultured marine bacterium Ant4D3; DQ295237 | 95.5 | Antarctic sea water | |
| Station2_20_08 | 1535 | Uncultured marine bacterium Ant4D3; DQ295237 | 97.6 | Antarctic sea water | |
| Station2_3_09 | 1526 | Uncultured marine bacterium Ant4D3; DQ295237 | 97.1 | Antarctic sea water | |
| Station2_8_09 | 1536 | Uncultured marine bacterium Ant4D3; DQ295237 | 95.5 | Antarctic sea water | |
| Station2_10_09 | 1536 | Uncultured Gammaproteobacterium; Arctic96A-14; AF354611 | 96.6 | Arctic Ocean (depth of 55m) | |
| Station2_12_09 | 1533 | Marine Gammaproteobacterium HTCC2188; AY386344 | 97.3 | Reference taxon 247644 | |
| Station2_14_09 | 1537 | Uncultured marine bacterium; KG_A3_120m15; EU005719 | 95.6 | Southern Ocean: Kerguelen Plateau (depth of 120m) | |
| Station3_1_08 | 1488 | Uncultured bacterium; S23_131; EF572032 | 96.2 | Costa Rica: Coco's Island site 23 | |
| Station3_9_08 | 1537 | Uncultured Gammaproteobacterium MS024-3A; EF202341 | 99.4 | USA: Maine, Boothbay Harbor (depth of 1m) | |
| Station3_11_08 | 1496 | Uncultured marine bacterium Ant4D3; DQ295237 | 99.6 | Antarctic sea water | |
| Station3_12_08 | 1527 | Uncultured proteobacterium OCS44; AF001650 | 98.9 | Reference taxon 62674 (related to the SAR86 cluster) | |
| Station3_13_08 | 1532 | Uncultured Gammaproteobacterium TAI-8-61; AM259782 | 95.6 | Adriatic Sea (sponge endosome) | |
| Station3_2_09 | 1246 | Glaciecolamesophila (T); KMM 241; AJ488501 | 91.3 | Reference taxon 197222 | |
| Bacteroidetes | Station1_12_09 | 1525 | Uncultured marine bacterium; KG_A3_120m24; EU005728 | 90.6 | Southern Ocean: Kerguelen Plateau (depth of 120m) |
| Station2_18_08 | 1512 | Uncultured marine bacterium; KG_A3_120m24; EU005728 | 99.6 | Southern Ocean: Kerguelen Plateau (depth of 120m) | |
| Station3_14_08 | 1510 | Uncultured Bacteroidetes bacterium; CF10; AY274847 | 98.6 | USA: Delaware River estuary | |
| Unclassified Proteobacteria | Station1_11_08 | 1533 | Uncultured Gammaproteobacterium; Arctic96B-1; AF353242 | 91.9 | Arctic Ocean (depth of 55m) |
| Station3_7_08 | 1506 | Uncultured marine bacterium; KG_C11_100m36; EU005922 | 92.8 | Southern Ocean: Kerguelen Plateau (depth of 100m) | |
| Station2_4_09 | 1512 | Colwelliasp. WED7.5; AY536568 | 95.0 | Reference taxon 264619 | |
| Station2_6_09 | 1478 | Uncultured marine bacterium Ant4D3; DQ295237 | 91.8 | Antarctic sea water | |
| Station1_14_09 | 1523 | Uncultured marine bacterium Ant4D3; DQ295237 | 93.2 | Antarctic sea water | |
| Station3_7_09 | 1539 | Uncultured marine bacterium Ant4D3; DQ295237 | 91.2 | Antarctic sea water |
Clone-ID: Station1, Station2 and Station3 means sampling site; number_08 or number_09 means clon number and sampling year. Closest relative accession number from RDP Database Project II Release 10.22; 1,418,497 sequences), and their 16S rRNA site isolation additional information.
Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 4 25 by using the Minimun Evolution method (Using the Maximum Composite Likelihood model). In the parsimony analysis, phylogenetic trees were constructed using the TNT program12. Here, we used multiple random addition sequences followed by tree bisection reconnection (RAS+TBR). Thus, heuristic searches were implemented using 1000 RAS, saving two trees per replication. Maximun Likelihood methods were also used and constructed by using PHYML13 (data not shown).
In order to assess the support for the identified groups, a bootstrap analysis (1000 replications) was performed.
Entropy calculationEntropy data were calculated by using BioEdit14. Sequence aligments from each site and date sampling were compared. The sum of entropy for all alignment sites was compared to asses space and time assemblage heterogeneity.
Nucleotide sequence accession numbersThe sequences determined in this study have been submitted to the GenBank database and assigned Accession Nos. JF927215-JF927274.
ResultsSite and date of isolation heterogeneity of the assemblagesThe heterogeneity of sequence collections (assessed by alignment entropy) showed that the most unvariable Shannon's entropy throughout time was observed in the open sea location (Station 3). Moreover, locations under the influence of both meltwater runoff and glacial ice (Station 1 and Station 2) were characterized by more variable bacterial assemblages with time, but with different trends. Sequence alignment entropy of bacterial assemblages in Station 1 was higher in 2009 than in 2008; conversely, Station 2 sequence alignment entropy was higher in 2008 than in 2009. Regarding the degree of heterogeneity in isolation sites, Station 3 showed the most heterogeneous assemblages in 2008 while Station 1 exhibited the most heterogeneous assemblages in 2009 (Data not shown).
Diversity of Antarctic free-living marine bacteria at Poter Cove, King George IslandFragment length polimorphism observed in marine samples obtained from Potter Cove waters near Jubany Station suggested high diversity during all the cloning experiment (range 1222 to 1539 nt). Moreover, EcoRI cut site was present in an average of 54.5% of the amplified fragments (maximun: 66.7%, minimun: 37.5%) for each site and time of sampling (see the selection criteria for sequenced clones).
All cloned sequences could be addressed to phyla of the domain Bacteria. Four phylogenetic groups of bacteria (affiliated by using RDP Database Project II and molecular phylogeny) dominated the PCR-based bacterial SSU rRNA gene clone library (Table 2). Members of the class Alphaproteobacteria accounted for 50% (30/60 clones) of the bacterial clone library whereas Gammaproteobacteria for 31.7% (19/60 clones). The class Betaproteobacteria and the phylum Bacteroidetes were almost equally represented in the library, accounting for 3.3% (2/60) and 5.0% (3/60) respectively. In addition, six out of 60 clones (10%) could not be classified by the RDP classifier tool (unclassified Proteobacteria). However, three out of six unclassified Proteobacteria were classified as Alphaproteobacteria and the remaining three sequences were classified as Gammaproteobacteria, respectively, when RDP Classifier confidence threshold was 85%.
Identity matrix of the sixty sequences from Potter Cove showed a mean value of 0.764 (SD: 0,095, maximum: 0.996, minimum: 0.431). In addition, when the sequences were compared with their closest relatives from the data bank, 40% of the sequences (24 out of 60) had sequence identities >99% compared to previously and most closely related sequences (Table 2); another 25% (15 out of 60 of the cloned sequences) had sequence identities that were between 96% and 99%, and finally, 35% (21 out of 60 of the cloned sequences) had sequence identities that were <96% compared to those previously described.
Sequences from the dominant class Alphaproteobacteria branched closely and were ascribed to six groups: a) order Rhodobacterales (16/30 clones, all of them included in the Rhodobacteraceae family but none could be identified to genus level); b) order Rickettsiales (8/30 clones, all of them belonging to the genus Pelagibacter, a member of the SAR11 clade); c) order Sphingomonadales (1/30 clones ascribed to genus Sphingomonas within the Sphingomonadaceae family); d) order Rhodospirillales (1/30 clones representing an unclassified member of the family Rhodospirillaceae); e) order Caulobacterales (1/30 clones, included within genus Asticcacaulis of the Caulobacteraceae family) and f) unclassified Alphaproteobacteria (3/30 clones).
Sequences affiliated to class Gammaproteobacteria were grouped into three groups: a) unclassified Gammaproteobacteria was the dominant group of sequences (17/19 clones); b) order Alteromonadales (1/19 clones belonging to the Pseudoalteromonadaceae family and the Pseudoalteromonas genus) and c) order Xanthomonadales (1/19 clones, affiliated to the Xanthomonadaceae family and the Dyella genus).
Sequences from members of phylum Bacteroidetes were affiliated to the order Flavobacteriales (2/3 clones, one of them belonging to genus Polaribacter into the family Flavobacteriaceae and the other one was an unclassified member of the order Flavobacteriales). The remaining Bacteriodetes (1/3) sequence could not be identified, not even at class level. On the other hand, one of the Betaproteobacteria sequences was ascribed to the Methylophilaceae family (1/2 clones) and the other was unclassified(1/2 clones). No Cyanobacteria were identified at all.
Phylogenetic analysis of Potter's Cove free-living Rhodobacteraceae SSU rRNA clonesThe 16/60 Rhodobacteraceae cloned sequences isolated from Potter Cove, near Jubany Station, were not associated with particulated organic matter, since they were collected from the fraction smaller than 1.5μm. Moreover, Rhodobacteraceae sequences from Potter Cove had a wide sequence identity matrix (data not shown) with an average of 90.9% (maximum: 99.8% and minimum: 72.7%).
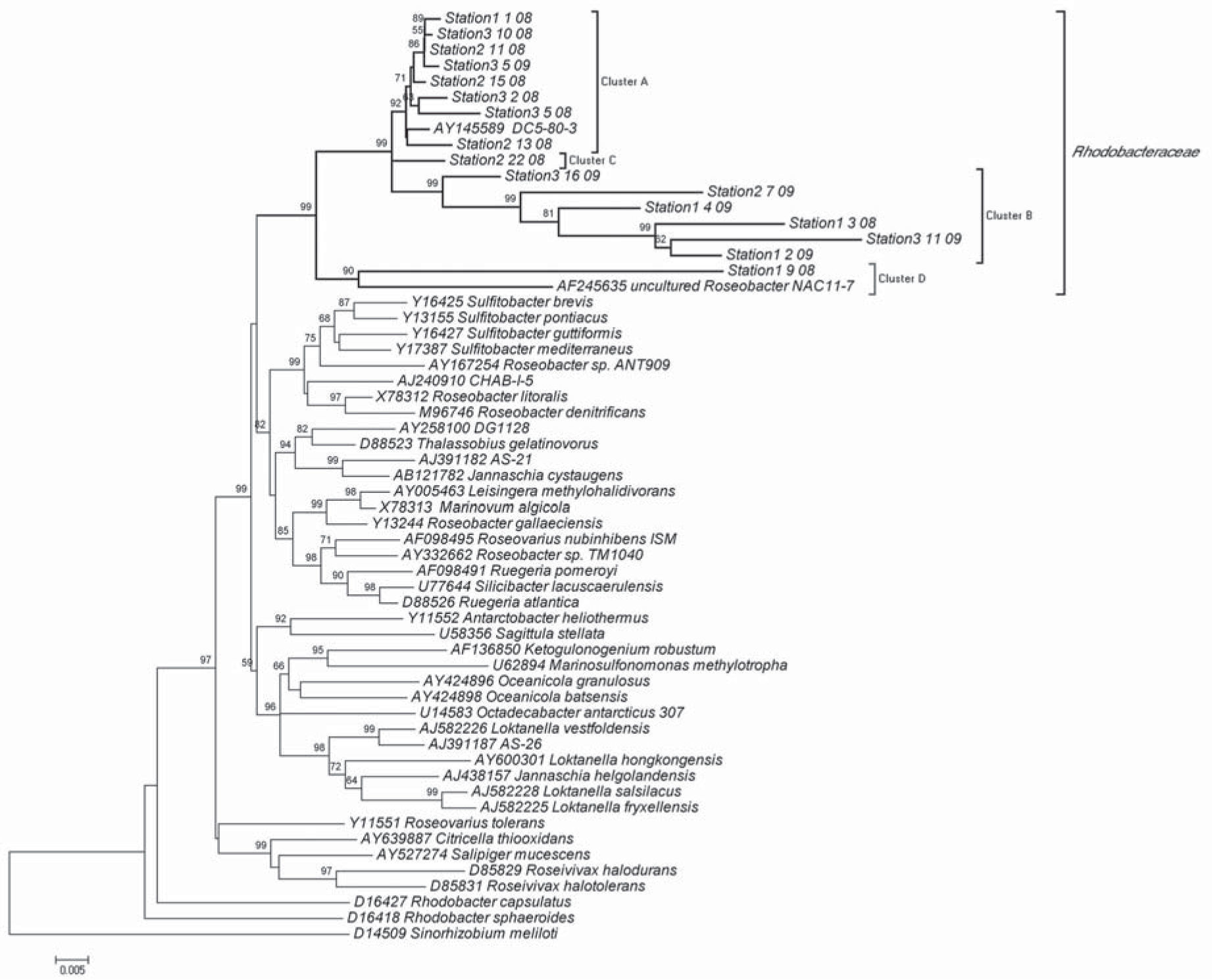
In order to conduct a more detailed phylogenetic analysis and a better description of the diversity within the planktonic members of the Rhodobacteraceae family, we compiled the same reference data set of Roseobacter 16S rRNA gene sequences previously described6. The results, using distance, Maximum Likelihood (ML) and Parsimony methods were similar; Minimum Evolution tree is depicted in Figure 1. The sixteen Rhodobacteraceae sequences from Potter Cove grouped into two of the clusters defined in Buchan's analysis.
. The tree was constructed using Mega 4 25 and the Minimun Evolution method. The bar represents Maximum Composite Likelihood evolutionary distances. Bootstrap values of >70% are shown at branch nodes (1000 iterations). S. meliloti (D14509) served as the outgroup.")
Phylogenetic relationship among sixteen Potter Cove Rhodobacteraceae sequences and a sequence set from Buchan et al., 2005. The tree is based on positions 92 to 1443 of the 16S rRNA gene (E. coli numbering system). The tree was constructed using Mega 4 25 and the Minimun Evolution method. The bar represents Maximum Composite Likelihood evolutionary distances. Bootstrap values of >70% are shown at branch nodes (1000 iterations). S. meliloti (D14509) served as the outgroup.
The first group of Potter Cove‘s free-living Rhodobacteraceae was closely related to the DC5-80-3 Octadecabacter-Ruegeria group (AY145589, uncultured Alphaproteobacterium isolated from the Weser estuary); however, this group could be sub-divided into two clusters. Cluster Rhodo-A was composed of eight out of sixteen Rhodobacteraceae sequences and was almost exclusively (7/8 sequences) composed of clones isolated in 2008, with 92% of bootstrap value. The other cluster, Rhodo-B, included six Rhodobacteraceae sequences, most of which (5/6) were clones isolated in 2009 (99% of bootstrap value). Finally, Station2 22 08 sequence collapsed at the Rhodo-B root. Noticeably, cluster Rhodo-B could also be splitted into several well supported sub-clusters, each containing distant members (see boostrap values and distance branch in Figure 1). Overall average genetic distance within cluster Rhodo-B was 4.5% (SD: 1.22%). In contrast, cluster Rhodo-A showed an overall average distance of 0,8% (SD: 0,46). The intergroup overall average genetic distance was considered extremely significant (ANOVA, Tukey-Kramer Multiple Comparisons Test, p=0,001).
Finally, the second group of Rhodobacteria (a single sequence, Station1_9_08) was distantly related to NAC11-7 and named Rhodo-C (AF245635, isolated from the North Atlantic algal bloom).
Potter Cove's Rhodo-B cluster has biogeographic identitySeveral features could be observed in the Rhodo-B group. i) five out of six sequences from cluster B (except for Station 3-16-09 sequence) exhibited a common nucleotide motif consisting of a 21 nucleotide insertion with no identical sequences (alignment position 1220-1240). This insertion is shared (both in alignment position and nucleotide length) with the unrelated Sinorhizobium meliloti (D14509) but not with any of Buchan‘s set of Roseobacter lineage sequences. ii) When the sixteen Rhodobacteraceae sequences from Potter Cove were compared with the 115 nearest neighbors using the RDP, sequences from cluster Rhodo-B did not group with any of the Rhodobacteraceae sequences isolated from Usuhaia seawater (data not shown)20. iii) Cluster Rhodo-B had a slightly lower G-C percentage than cluster Rhodo-A (53% versus 53.7%). All the above commented features suggested that 16S RNA sequences from cluster Rhodo-B have a specific nucleotide composition that gives them biogeographical identity.
In contrast, sequences from cluster Rhodo-A exhibited a short genetic distance regarding the closest relatives from the data bank, which showed to be broadly intermingled with sequences from a lot of geographical sites in the phylogenetic tree (data not shown).
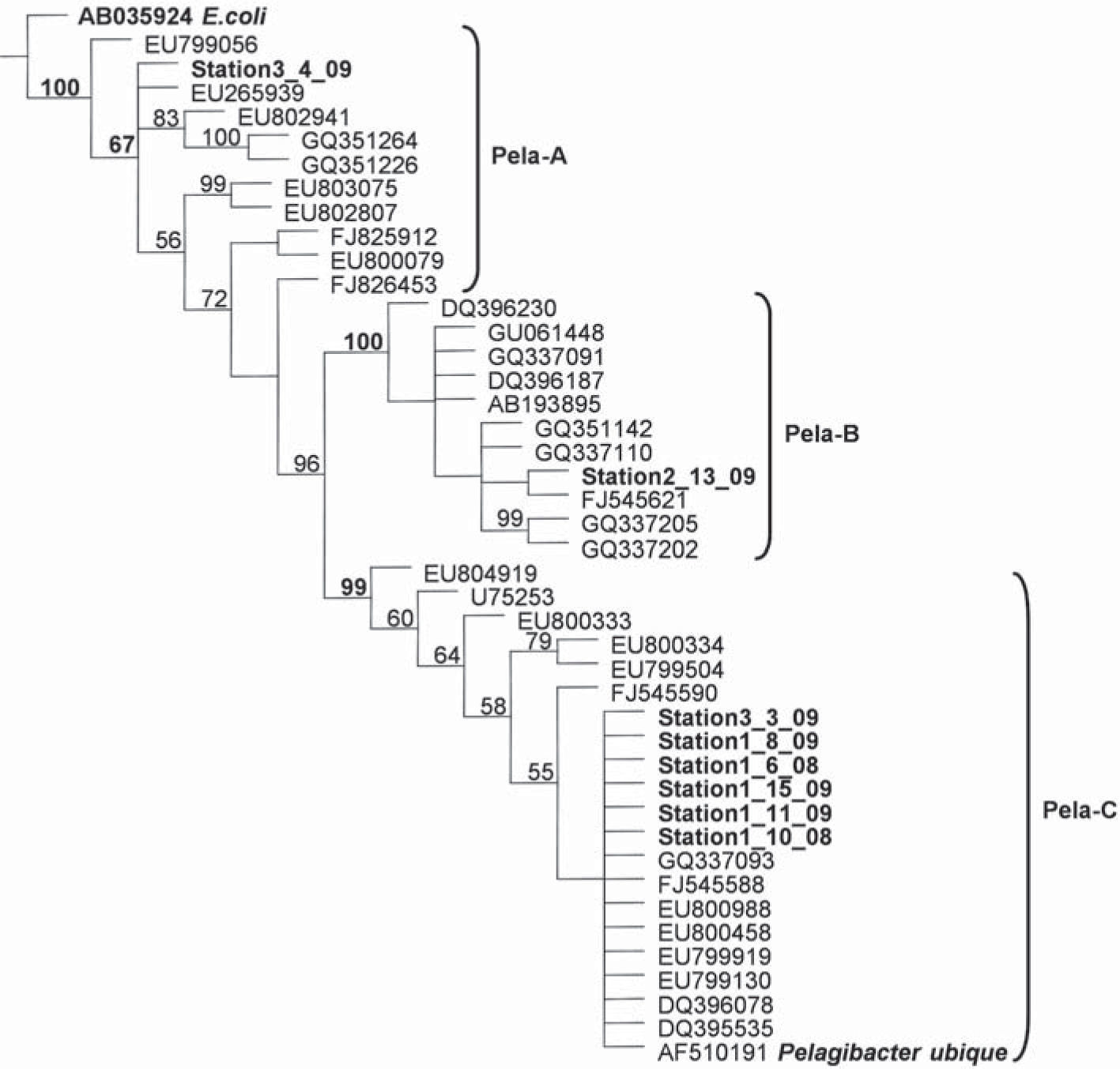
Pelagibacter lineage in Antartic coastal sea waterEight Potter Cove's Pelagibacter isolated sequences were analized together with their 35 nearest neighbors sequences from RDP Database Project II by the Parsimony method (Fig. 2). Minimum Evolution and Maximun Likelihood methods were also used and the topology tree was similar (data not shown). The phylogenetic tree depicted three groups of sequences referred to as Pela-A, Pela-B and Pela-C. The first most ancestral and not well supported group, Pela-A, comprised eleven sequences and included Station3-4-09 as a collapsed to root member of the group. The closest relative to Station 3-4-09 was a sequence (EU265939) obtained from Nitinat lake in Canada at a depth of 20m. The remaining closest relatives of the Pela-A group included some sequences obtained from deep sea water (3336 meters-depth near Panama and 1000 meters-depth from the North East Pacific Ocean). The second group, Pela-B, was a very well supported group (bootstrap 100%) of eleven sequences that included the Station2-13-09 clone, whose closest relative was a sequence isolated from the Northern Yellow Sea (FJ545621). Other close relatives were sequences obtained from 400-1000 meter-depth in the Arctic Sea. The third group, Pela-C, was another well supported group (bootstrap 99%) of twenty one sequences that included six out of eight Potter Cove's Pelagibacter sequences. These sequences were closely related to Pelagibacter ubique HTCC1062 (AF510191)
Rich assemblage of marine Gammaproteobacteria isolated near Jubany Station served as the outgroup.")
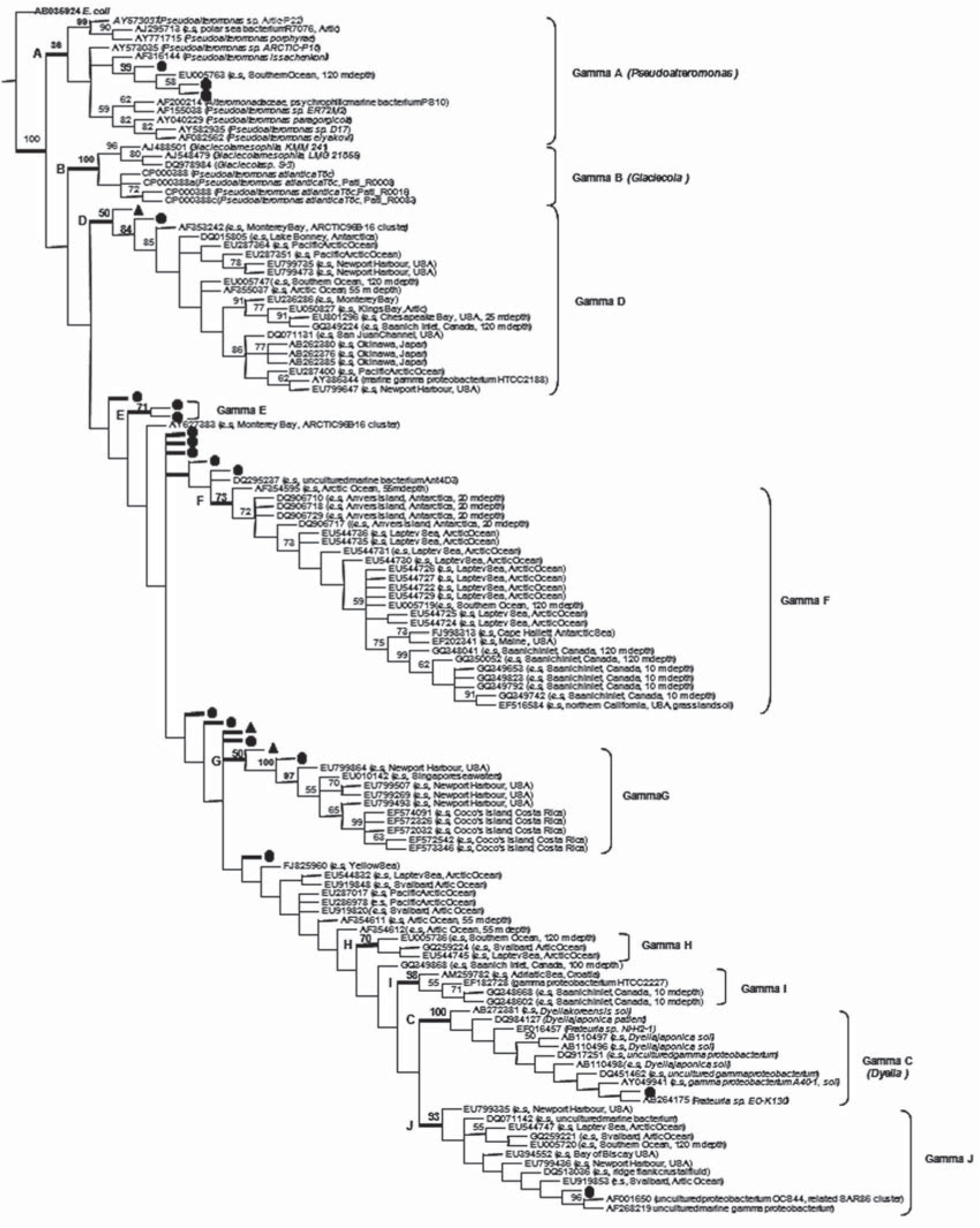
As most of the Potter Cove sequences that were identified as Gammaproteobacteria using the RDP Classifier (having a 95% confidence threshold) could not be adequately classified at genus level, a Parsimony phylogenetic tree was constructed to analyze their relationship with one hundred twelve closest relatives uploaded from data banks (Fig. 3). In this analysis, three sequences which were classified as Gammaproteobacteria by RDP Classifier only when using a 85% confidence threshold (Station 1-11-08, Station 2-6-09 and Station 1-14-09) were also included. Then, Gammaproteobacteria from Potter Cove and their closest relatives determined ten clusters (Gamma A to Gamma J) and a set of ungrouped sequences. Results showed that: i) Clusters Gamma A-C included those Gammaproteobacteria that were classified as Pseudoalteromonas, Glaciecola and Dyella genus by the RDP Classifier. ii) Clusters Gamma D-J comprised only unclassified environmental sequences grouped with bootstrap values equal or above 50%. iii) A set of Potter Cove and other environmental isolates, which included the remaining unclassified sequences, were not grouped under the previous criteria.
 identifies Potter Cove Gamma-Proteobacteria (RDP Classifier confidence threshold of 95%) and symbol (▲) identifies Potter Cove Gamma-Proteobacteria (RDP Classifier confidence threshold of 80%). E.coli (AB035925) served as the outgroup.")
Parsimony phylogenetic tree of Potter Cove Gamma-Proteobacteria sequences and their one hundred twelve closest relatives. Symbol (●) identifies Potter Cove Gamma-Proteobacteria (RDP Classifier confidence threshold of 95%) and symbol (▲) identifies Potter Cove Gamma-Proteobacteria (RDP Classifier confidence threshold of 80%). E.coli (AB035925) served as the outgroup.
Three of the Potter Cove sequences were included within cluster Gamma A (Pseudoalteromonas genus) by the Parsimony method (Station1-5-09, Station 2-17-08 and Station 3-2-09), while the RDP Classifier only identified Station 1-5-09 sequence as member of the genus Pseudoalteromonas. These three sequences were closely related among themselves, with a sequence previously obtained from the Southern Ocean (EU005763, bootstrap value 99%)29. Despite including in this analysis some sequences corresponding to members of the genus Glaciecola within the closest relatives, no Gammaproteobacteria sequences from Potter Cove grouped phylogenetically with them. Sequences ascribed to the Dyella genus were well supported in the tree (bootstrap value 100%); this tree branch included sequences obtained from soil samples and also from our marine sequence Station2-10-08.
Cluster Gamma D (bootstrap value 50%) included two sequences from Potter Cove along with many environmental ones. All these environmental sequences were obtained from different marine sites and comprised uncultured bacteria AF353242 belonging to the ARCTIC96B-16 cluster2, as the most related one.
Cluster Gamma E only included two sequences from this work whereas cluster Gamma F comprised well suported sequences in the tree (bootstrap value 73%) that were almost exclusively from polar environments (with the exception of EF516584). However, although not well supported, they were related to the ancestral Station 2-20-08 and Station 1-22-08 from this study.
Cluster Gamma G grouped sequences from warm and temperate marine sites together with Station 2-6-09 and Station 3-1-08 from this work as the most ancestral members of this group.
Clusters Gamma H and Gamma I included no Potter Cove‘s Gammaproteobacteria sequences and cluster Gamma J, a very well supported group (bootstrap value 93%), contained sequences from different marine latitudes and depths together with Station3-12-08 sequence, which was closely related to an uncultured proteobacterium OCS44 (AF001650) related to the SAR86 cluster22.
Finally, because they were collapsed to root or not supported by bootstrap value, the sequence set that did not group in any cluster included the remaining eleven Gammaproteobacteria sequences from Potter Cove which had not close relatives.
DiscussionStudies on the bacterial diversity of seawater from cold regions such as west Antarctica based on the 16S rDNA sequencing and phylogenetic approach are limited17,18. The present study provides the first report on bacterioplankton assemblages and several potential new species at Poter Cove, King George Island, South Shetland Islands. The coastal zone of Potter Cove is considered a Specially Protected Area (SPA) because of its diverse and extensive vegetation and fauna, which constitutes a representative sample of the ecosystem of the Antarctic Peninsula. For this reason and also due to the presence of the Jubany Scientific station in the area, whose activity could represent a relevant factor affecting the normal bacterial community structure, a deeper knowledge of the present status of such communities is urgently required.
Results of entropy variation as a function of time for bacterial assemblages from Station 1 and Station 2 (which are frozen in winter and thaw in summer) evidenced the sensitivity of the Antarctic environment to this climatic factor. Station 3, which remains unfrozen all around the year, showed a set of more stable and rich microorganisms over time, as was suggested by their entropy values.
The identity matrix of sixty sequences obtained in this study indicated that the seawater bacterioplankton assemblages studied here exhibit great diversity. Furthermore, 35% of the cloned sequences (21 out of 60) could be new isolates due to their low identity value (< 96%) with their closest relatives. In addition, most cloned sequences were more closely related to uncultured, environmental sequences. These two observations highlight the scarce knowledge of the composition of Antarctic marine bacterial assemblages and also suggest that the bulk of sequences obtained from the total DNA isolated from the seawater did not match any previously cultured and described bacterial strain.
The high predomince of 16S rDNA-amplified sequences of Phylum Proteobacteria (50% of Alphaproteobacteria and 31,7% of Gammaproteobacteria) was also reported from seawater samples collected off Ushuaia, Argentina, Sub-Antarctica20 and other oceanic areas of the Southern oceans18. However, this predominance could not be associated with Antarctic and subAntarctic seawaters because it was reported for the Arctic2 and also for temperate or warm coastal marine waters9,19.
Despite the few sequences obtained, the Rhodobacteraceae component of Potter Cove bacterioplankton showed several remarkable features. Cluster Rhodo-A seems to be a cosmopolitan genotype because of their narrow intra-group genetic distance when compared with 115 closest relatives from the data bank. Conversely, cluster Rhodo-B seems to be an endemic, biogeographically splitted and genetically divergent genotype within the Rhodobacteracea family. This result suggests that it could be a different Rhodobacteracea lineage exclusive of the areas in the south of the Polar Front. This fact, which was also described from a transect made in another geographic longitude of the polar front24, suppresses the question regarding the cosmopolitan or endemic character of planktonic species. Although some authors have proposed that marine planktonic microorganisms should be cosmopolitan and that endemic species should be rare10,11, our results support the hypothesis of the existence of some degree of endemism. Such endemism could be related to a marked latitudinal gradient of OTU richness, as was stated in a study of global patterns of diversity in marine bacterioplankton19. For Antarctic marine waters, it could be possible that the Polar front, which represents a strong 25 million-year old barrier to free latitudinal exchange of water, be strongly contributing to generate a distinctive biogeographical discontinuity, as was previously suggested7. This role of the polar front as a barrier to the spreading of some species has been also proposed to explain the geographical distribution of two different phylotypes of the Roseobacter clade, one north and one south of the front24. In addition, the fact that the bulk of sequences of cluster Rhodo-A and Rhodo-B were isolated in summer (seven out of eight sequences from summer 2008 and five out of six sequences from summer 2009, respectively), might be indicating a temporal segregation of these isolates. This might be caused by a possible inter-annual change in the thawing of Fourcade Glacier (which surrounds Jubany Station and could be acting as a store of fossil microorganisms).
There is still scarce information available on the abundance and phylogenetic affiliation of the SAR11 clade in west Maritime Antarctica, where Potter Cove is located. Most of the SAR 11 isolates from this study (six out of eight clones) were closely phylogenetically grouped with Pelagibacter ubique, the first cultured member of the SAR11 clade and isolated from cold Oregon coastal waters. This suggests the existence of a cosmopolitan SAR11 lineage in Potter Cove. Remarkably, this cosmopolitan lineage of SAR11 was preferentially isolated from Station 1 (inner part of Potter Cove). On the other hand, the two remaining clones (Station 2-13-09 and Station 3-4-09) were grouped with some environmental isolates from very deep sea water from different parts of the world, beside the fact that our Pelagibacter sequences were obtained from surface sea water. Although no inference in this respect can be made from the results obtained in this work, it is interesting to take into account for future researches the fact that Antarctic bottom waters could be responsible for an effective transport of psychrophilic microorganisms from Antarctic shallow waters to temperate and even tropical deep sea basins, as was proposed several years ago in a study of sediments from the Sierra Leone Abyssal Plain23.
With regard to Gammaproteobacteria from Potter Cove, our results suggest that, except in the case of those belonging to genus Pseudoalteromonas and Dyella (Station 1-5-09 and Station 2-10-08, respectively), the rest of the sequences could belong to microorganisms not yet described in the database, despite the fact that RDP release 10, update 22 consists of 1,418,497 aligned and annotated 16S rRNA sequences. Moreover, these results could suggest that unclassified Potter Cove Gammaproteobacteria are endemic of this extreme and poorly described geographical area. Ancestry of most of the Gammaproteobacteria isolates described in this study could suggest the input of “old microorganisms” from the defrosting of the Fourcade Glacier (which has retreated one kilometer from its original front line in the last 15 years).
Finally, we highlight the absence of Cyanobacteria sequences in our samples. This observation is not related to a bias in the primer annealing, because when we blasted the used primers in the databank, it did not show any bias against Cyanobacteria. Although it is known that Cyanobacteria are not as abundant in polar oceans as in other marine areas or even in polar freshwater systems26, the fact that we have not obtained cyanobacterial sequences at all, reflects the scarce abundance of this group in Potter Cove and also opens several interesting ecological questions such as who takes the role of cyanobacteria as primary producer in this system. Future answers to this and other related questions will contribute to a better undestanding of Potter Cove and other similar Antarctic marine coastal water ecosystems.
In conclusion, we here report a rich sequence assemblage composed of most of the isolates that are widely divergent among themselves and between the most closely related sequences currently deposited in the databank. Geographical isolation of the polar front could explain these results as reported24. Furthermore, genetic divergence of both unclassified Alphaproteobacteria and Gammaproteobacteria sequences described here highlights the relevance of future studies evaluating the magnitude of microbial input caused to marine microbial communities by glacier defrosting in Potter Cove and other similar Antarctic coastal areas as well.
Ethical disclosuresProtection of human and animal subjects.The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of data.The authors declare that no patient data appear in this article.
Right to privacy and informed consent.The authors declare that no patient data appear in this article.
FundingThis work was supported by a grant from Universidad de Buenos Aires (SECyT-UBA, B417).
Conflicts of interestThe authors declare that they have no conflicts of interest.
