




La podocina es una proteína localizada en el diafragma de filtración glomerular donde participa en la regulación de la filtración glomerular. Las mutaciones del gen NPHS2, que codifica a la podocina, son la principal causa de síndrome nefrótico corticorresistente (SNCR) autosómico recesivo en niños.
ObjetivosIdentificar mutaciones de NPHS2 en niños chilenos con SNCR, y establecer la prevalencia de las variantes más frecuentes en un grupo de adultos sanos.
Pacientes y métodoAnálisis mutacional de NPHS2 en 34 niños chilenos con SNCR. Una vez identificadas las dos variantes de NPHS2 de mayor frecuencia, se realizó un screening de estas mutaciones en 223 adultos sanos. El análisis mutacional se realizó por secuenciación directa de los ocho exones codificantes amplificados por reacción de polimerasa en cadena. La secuenciación del DNA se realizó mediante método fluorométrico y las secuencias fueron evaluadas con el software SeqPilot. La asociación entre la presencia de variantes de NPHS2 y SNCR se calculó comparando las frecuencias alélicas entre los pacientes con SNCR y los voluntarios sanos utilizando prueba exacta de Fisher. Se consideró significativo p<0,05.
ResultadosSe detectaron mutaciones patogénicas de NPHS2 en siete de los 34 pacientes (21%) estudiados, de los cuales seis resultaron heterocigotos para p.R229Q y p.A284V. En voluntarios sanos la prevalencia de p.R229Q fue de 2,46%.
ConclusionesEste estudio muestra que p.R229Q y p.A284V son las variantes de NPHS2 más frecuentes en niños chilenos con SNCR. Por primera vez se describe esta asociación en niños chilenos, en base a la cual es posible proponer una estrategia de screening para estudio genético en pacientes con SNCR y sus familias. Se propone una estrategia de búsqueda de p.R229Q y p.A284V en forma paralela o secuencial en estos pacientes.
Podocin is a protein located in the glomerular slit diaphragm where it takes part in the regulation of glomerular filtration. Mutations of the NPHS2 gene that codes podocin are the main cause of autosomal recessive steroid resistant nephrotic syndrome (SRNS).
ObjectivesTo identify the NPHS2 mutations in Chilean children with SRNS, and to determine the prevalence of the most common variants in a group of healthy adults.
Patients and methodsMutation analysis of NPHS2 in 34 Chilean children with SRNS. Once the two most common variants of NPHS2 were identified, screening for these mutations was performed on 233 healthy adults. The mutation analysis was performed by the direct sequencing of the eight coding exons by polymerase chain reaction amplification. The DNA sequencing was performed using a fluorometric method, and then evaluated with SeqPilot™ software. The relationship between the presence of NPHS2 variants and SRNS was calculated by comparing the allele frequency between patients with SRNS and those of the healthy volunteers using the exact Fisher test. A P<.05 was considered significant.
ResultsPathogenic NPHS2 mutations were detected in 7 (21%) of the 34 patients studied, of which 6 were heterozygotes for p.R229Q and p.A284V. The presence of p.R229Q was 2.46% in the healthy volunteers.
ConclusionsThis study shows that p.R229Q and p.A284V are the most frequent variants in Chilean children with SRNS. It is the first time that this relationship has been reported in Chilean children. Based on this, a screening strategy is proposed for the genetic study in patients with SRNS and their families. A parallel or sequential search strategy for p.R229Q and p.A284V in these patients is proposed.
El síndrome nefrótico (SN) se define como la asociación de proteinuria (>40mg/m2/h), hipoalbuminemia (<2,5mg/dl) y la presencia de edema. La hiperlipidemia anteriormente se consideraba criterio diagnóstico, pero esta es secundaria a la hipoalbuminemia1. La incidencia anual de SN idiopático en niños en Estados Unidos y Europa se ha estimado en 1-3 por 100.000 niños, con una prevalencia acumulada de 16 por 100.000 niños. En base a la respuesta a la terapia corticoesteroidal, se ha dividido en dos categorías, los que responden a la terapia corticoidal: SN córticosensible, que incluye el SN corticodependiente y el SN recaedor frecuente y SN recaedor infrecuente; y los que no responden al tratamiento con corticoides: corticorresistente (SNCR)1,2. Aproximadamente 10-20% de los niños con SN idiopático no logran remisión con el tratamiento con corticoides2. Mientras que la mayoría de los casos con SNCS muestra en la biopsia renal enfermedad de cambios mínimos (ECM), los casos de SNCR presentan principalmente glomeruloesclerosis focal y segmentaria (GEFS) o esclerosis mesangial difusa3,4. El SNCR representa una entidad clínica desafiante para el nefrólogo infantil, dado que el 50% de los niños progresan a la enfermedad renal terminal dentro de los 15 años5.
En los últimos años se han identificado mutaciones en genes que codifican proteínas del podocito en varias formas de SNCR3. Dentro de estos, mutaciones del gen NPHS2, que codifica la proteína podocina, representa la causa más frecuente de SNCR autosómico recesivo en niños4. La podocina es una proteína localizada en el diafragma de filtración glomerular, donde participa en la organización estructural y en la regulación de la filtración glomerular4. Por medio del clonamiento posicional, Boute1 logró encontrar una nueva proteína específica del podocito, la podocina, codificada por NPHS2 en el cromosoma1q25-31 y que se asociaba con formas autosómicas recesivas de SNCR. La podocina es una proteína de membrana del podocito de 383 aminoácidos, tiene una estructura similar a una horquilla y se expresa exclusivamente en el diafragma de filtración glomerular. Después de la descripción inicial de NPHS2, se han identificado numerosas variantes del gen en diversos grupos raciales y étnicos de pacientes con SNCR, incluidas más de 30 mutaciones patogénicas y algunos polimorfismos6. Podocina es la responsable de la mayoría de los SNCR infantiles, el 42% corresponden a formas familiares y 10% de los casos esporádicos; también cabe destacar que se ha encontrado en el 39% de los pacientes con síndrome nefrótico congénito.
Los escasos estudios de mutaciones de NPHS2 realizados en individuos chilenos con SNCR7–9 muestran exclusivamente pacientes heterocigotos para el polimorfismo p.R229Q y la mutación p.A284V, similar a lo reportado en población española10. Los estudios existentes corresponden principalmente a reportes de casos, tanto adultos como niños. No existen estudios diseñados para conocer las variantes de NPHS2 y su frecuencia en niños chilenos con SNCR ni en un grupo de voluntarios sanos.
Los objetivos de este estudio son identificar mutaciones de NPHS2 en niños chilenos con SNCR, y establecer la prevalencia de las variantes más frecuentes en un grupo de adultos sanos de la población general.
Pacientes y métodoEstudio descriptivo de corte transversal. Criterios de inclusión: pacientes en control en la Unidad de Nefrología del Hospital Luis Calvo Mackenna entre los años 2009 y 2011, con el diagnóstico de SNCR, que hayan firmado el consentimiento informado y/o asentimiento. Criterios de exclusión: pacientes en que se logró la remisión del cuadro con el uso de terapia inmunosupresora, pacientes con SN de causa secundaria y recurrencia postrasplante. El estudio fue aprobado por el Comité de Etica del Hospital Luis Calvo Mackenna.
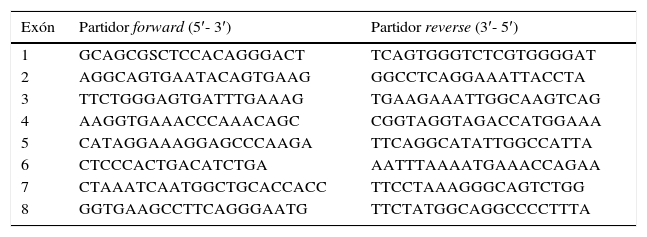
Se realizó análisis mutacional de NPHS2 en 34 niños con diagnóstico de SNCR, se obtuvo DNA genómico desde sangre periférica utilizando un método estándar (QIAamp DNA Mini and Blood Mini, Qiagen) según protocolo del fabricante. Se realizó análisis mutacional de NPHS2 mediante secuenciación directa de los ocho exones codificantes amplificados por reacción en cadena de la polimerasa (PCR) utilizando partidores de acompañamiento intrónico. La secuencia de los partidores se obtuvo de datos publicados previamente (tabla 1)11. La secuenciación del DNA se realizó mediante método fluorométrico (Big-Dye Terminator Sequencing Kit; ABI 3700 DNA sequencer, Applied Biosystems, Foster City, CA, EE. UU.) de acuerdo a protocolo del fabricante, y las secuencias fueron evaluadas con el software SeqPilot (JSI Medical Systems GmbH Kip penheim, Alemania). Este proceso se llevó a cabo en el Laboratorio de Nefrología Infantil de la Universidad de Heidelberg, Alemania.
Partidores utilizados para el análisis mutacional de NPHS2
| Exón | Partidor forward (5′- 3′) | Partidor reverse (3′- 5′) |
|---|---|---|
| 1 | GCAGCGSCTCCACAGGGACT | TCAGTGGGTCTCGTGGGGAT |
| 2 | AGGCAGTGAATACAGTGAAG | GGCCTCAGGAAATTACCTA |
| 3 | TTCTGGGAGTGATTTGAAAG | TGAAGAAATTGGCAAGTCAG |
| 4 | AAGGTGAAACCCAAACAGC | CGGTAGGTAGACCATGGAAA |
| 5 | CATAGGAAAGGAGCCCAAGA | TTCAGGCATATTGGCCATTA |
| 6 | CTCCCACTGACATCTGA | AATTTAAAATGAAACCAGAA |
| 7 | CTAAATCAATGGCTGCACCACC | TTCCTAAAGGGCAGTCTGG |
| 8 | GGTGAAGCCTTCAGGGAATG | TTCTATGGCAGGCCCCTTTA |
Fuente: Boute et al.11
Se realizó búsqueda de las variantes p.R229Q y p.A284V de NPHS2 en 223 individuos sanos que acudieron como donantes voluntarios al Banco de Sangre del Hospital Luis Calvo Mackenna durante el año 2011. Se excluyeron aquellos casos que presentaran alguna enfermedad renal o que fueran familiares de los pacientes con SNCR. Se realizó búsqueda de las variantes p.R229Q y p.A284V en los voluntarios sanos, mediante amplificación por PCR del segmento de NPHS2 utilizando partidores publicados previamente (tabla 2)12. Posteriormente se llevó a cabo análisis de polimorfismos de longitud de fragmentos de restricción13–15 (ensayo PCR-RFLP), mediante digestión de productos de PCR utilizando las enzimas de restricción ClaI y HhaI (New England BioLabs) para p.R229Q y p.A284V respectivamente, según protocolo del fabricante. Los productos de digestión se visualizaron mediante electroforesis en gel de agarosa.
Partidores utilizados para el screening de las variantes p.R229Q y p.A284V
| Variante | Partidor forward (5′- 3′) | Partidor reverse (3′- 5′) |
|---|---|---|
| p.R229Q | AGGATTTACCACAGGATTAAGTTGTG | GGTAGGGAAACCCTCGAGTATCGAT |
| p.A284V | GCACACTCTGGTCACTCCAA | CGTTGAGGAGAGGGAGAGAC |
Fuente: Tsukaguchi et al.12
La asociación entre la presencia de variantes de NPHS2 y SNCR se calculó comparando las frecuencias alélicas entre los pacientes con SNCR y los voluntarios sanos utilizando prueba exacta de Fisher. Se consideró significativo p<0,05.
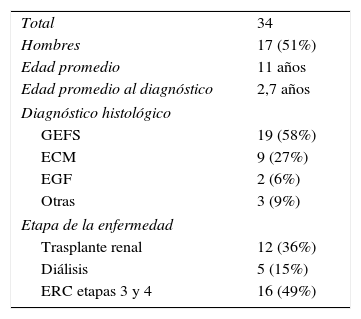
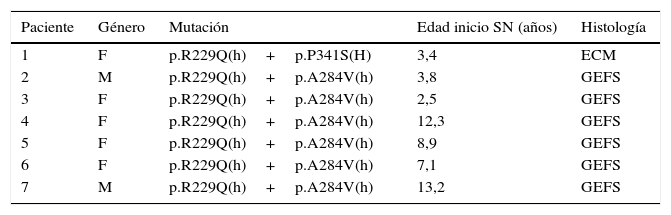
ResultadosLas características clínicas y epidemiológicas de los 34 pacientes con SNCR estudiados se muestran en la tabla 3. Se detectaron mutaciones patogénicas de NPHS2 en estado homocigoto o heterocigoto compuesto en siete de los 34 pacientes (21%) estudiados (tabla 4). Se identificaron dos variantes de NPHS2, p.R229Q, y p.A284V. Siete niños resultaron heterocigoto compuesto para p.R229Q y p.A284V. La frecuencia alélica de p.R229Q en los pacientes con SNCR fue de 10,29% (7/68 cromosomas) y la de p.A284V de 10,29% (7/68 cromosomas).
Características clinicoepidemiológicas de los pacientes con SNCR
| Total | 34 |
| Hombres | 17 (51%) |
| Edad promedio | 11 años |
| Edad promedio al diagnóstico | 2,7 años |
| Diagnóstico histológico | |
| GEFS | 19 (58%) |
| ECM | 9 (27%) |
| EGF | 2 (6%) |
| Otras | 3 (9%) |
| Etapa de la enfermedad | |
| Trasplante renal | 12 (36%) |
| Diálisis | 5 (15%) |
| ERC etapas 3 y 4 | 16 (49%) |
ECM: enfermedad de cambios mínimos; EGF: esclerosis global focal; ERC: enfermedad renal crónica; GEFS: glomérulo-esclerosis focal y segmentaria.
Pacientes con SNCR portadores de mutación en NPHS2
| Paciente | Género | Mutación | Edad inicio SN (años) | Histología |
|---|---|---|---|---|
| 1 | F | p.R229Q(h)+p.P341S(H) | 3,4 | ECM |
| 2 | M | p.R229Q(h)+p.A284V(h) | 3,8 | GEFS |
| 3 | F | p.R229Q(h)+p.A284V(h) | 2,5 | GEFS |
| 4 | F | p.R229Q(h)+p.A284V(h) | 12,3 | GEFS |
| 5 | F | p.R229Q(h)+p.A284V(h) | 8,9 | GEFS |
| 6 | F | p.R229Q(h)+p.A284V(h) | 7,1 | GEFS |
| 7 | M | p.R229Q(h)+p.A284V(h) | 13,2 | GEFS |
ECM: enfermedad de cambios mínimos; F: femenino; GEFS: glomérulo-esclerosis focal y segmentaria; (h): heterocigoto; (H): homocigoto; M: masculino; SN: síndrome nefrótico.
Las características epidemiológicas de los donantes voluntarios se muestran en la tabla 5. Se encontró la variante p.R229Q en 11 de los 223 voluntarios sanos de la población general, todos en estado heterocigoto, lo que significa una frecuencia alélica en este grupo de 2,46% (11/446 cromosomas). Este polimorfismo resultó ser significativamente más frecuente en el grupo de pacientes que en el grupo de individuos sanos (10,29 versus 2,46%), lo que indica una asociación de p.R229Q con SNCR (odds ratio=4,17; P=0,005). La mutación p.A284V no se encontró en este grupo.
DiscusiónPor primera vez en nuestro país se demuestran las mutaciones asociadas a SNCR en pacientes pediátricos.
Los resultados de este estudio muestran que las mutaciones de NPHS2 son una causa importante de SNCR en niños chilenos, similar a lo reportado en otras poblaciones4,16,17. En nuestro grupo de niños con SNCR, el 21% de los pacientes estudiados presentó una mutación patogénica de NPHS2 en estado heterocigoto compuesto. Las variantes más frecuentes corresponden al polimorfismo p.R229Q y a la mutación patogénica p.A284V, identificadas en estado heterocigoto compuesto en siete pacientes portadores de SNCR. Esto es similar a lo reportado previamente en pacientes con SNCR de Chile, Argentina y España7–10.
Se han descrito diversas variantes de NPHS2 en cohortes de pacientes con SNCR y controles sanos de diverso origen étnico. La variante no sinónima reportada más frecuentemente es p.R229Q6. En este estudio se encontró una frecuencia alélica para este polimorfismo de 2,46% en el grupo de voluntarios sanos, similar a lo reportado en la literatura1,9,16,18,19. La mutación p.A284V no se encontró en ninguno de los 223 voluntarios sanos, lo que coincide con lo descrito previamente en población europea y sudamericana1,9, se podría mejorar la representatividad del estudio teniendo controles sanos de todo el país.
Algunos autores han descrito que el suero de pacientes con GEFS aumenta la permeabilidad glomerular a la albúmina al incubar los glomérulos en ella in vitro. A diferencia de las formas inmunológicas del SN, estudios recientes muestran que defectos estructurales heredados de la barrera de filtración glomerular son responsables de una gran proporción de casos de SNCR, en la cual el podocito tiene un rol crucial en la patogénesis de la glomerulopatía10. Las mutaciones de NPHS2 fueron descritas inicialmente en SNCR de inicio precoz11 y estudios sucesivos definieron el fenotipo asociado a mutaciones de este gen, mostrando que los pacientes usualmente desarrollan SN antes de los 6 años de edad, presentan en su mayoría GEFS, no responden a terapia inmunosupresora y presentan menor riesgo de recidiva después del trasplante renal1,4,20,21. En forma paralela se describió la variante p.R229Q como un polimorfismo de la podocina frecuente en la población general, e inicialmente no se le atribuyó un efecto patogénico. Sin embargo, en base a estudios in vitro12 y poblacionales6,9,10,12 se ha propuesto un rol patogénico de p.R229Q describiéndose que los pacientes portadores de esta variante en estado heterocigoto compuesto con una mutación patogénica presentan SNCR de inicio tardío, después de los 18 años de edad. En este estudio reportamos seis pacientes heterocigotos compuestos para p.R229Q y la mutación patogénica p.A284V, todos con GEFS, y que llamativamente comenzaron con SNCR a edades más tempranas que lo descrito en la literatura, lo que abre la interrogante respecto a la existencia de determinantes locales, genéticos o ambientales, responsables de este fenómeno. Por otro lado, los resultados de este estudio muestran que p.R229Q fue significativamente más frecuente entre los pacientes en comparación con el grupo de donantes sanos, lo que es concordante con un rol patogénico de p.R229Q en el desarrollo de SNCR.
El polimorfismo p.R229Q presenta una distribución alélica desigual en las diferentes poblaciones en que se han estudiado, observándose con mayor frecuencia en personas de ascendencia europea1,16,22, y menos frecuentemente en descendientes africanos y asiáticos23–25. El análisis de haplotipos de este alelo mostró que está presente en un haplotipo frecuente en pacientes con SNCR. Estos datos sugieren que la variante p.R229Q surgió en un ancestro común, posiblemente en Europa, y se propagó posteriormente con la expansión de la población10,12. De igual forma, el análisis de haplotipos de la mutación p.A284V mostró que está presente en un haplotipo conservado en pacientes españoles con SNCR10. Debido a que p.A284V se ha detectado principalmente en pacientes españoles y de Sudamérica, es probable que la variante haya sido introducida por un colonizador español en la población hispánica26. Esto explicaría la alta frecuencia de pacientes portadores de estas dos variantes en este estudio y en estudios previos realizados con pacientes chilenos y argentinos7–9.
Además del polimorfismo p.R229Q, en este estudio se detectó una mutación con pérdida de sentido en los pacientes con SNCR, p.A284V. La que ha sido reportada previamente y se reconoce su rol patogénico9,10,27.
Este estudio incluyó 34 pacientes, lo cual representa a los pacientes que son derivados al Hospital Luis Calvo Mackenna, una proporción importante del total de pacientes con SNCR en Chile. Con el objetivo de conocer la realidad nacional de mutaciones en niños con SNCR se debería realizar un estudio colaborativo de las unidades de nefrología infantil de nuestro país.
Nuestros hallazgos muestran que en este grupo de niños chilenos con SNCR, las mutaciones de NPHS2 son una causa importante de la enfermedad. De acuerdo a esto, se podría implementar una estrategia de screening para buscar mutaciones de NPHS2 en pacientes con SNCR y sus familias. A partir de los datos obtenidos en este estudio, proponemos una estrategia consistente en la búsqueda de p.R229Q y p.A284V, en forma paralela, o bien secuencial, comenzando por p.R229Q y en caso de encontrarse presente, continuar con la búsqueda de p.A284V, especialmente en pacientes con ascendencia española. Si p.A284V es negativo, entonces se debería secuenciar el gen completo. El diagnóstico genético significaría un gran avance en lo concerniente al consejo genético en familias con mutaciones de NPHS2, puesto que esta información permitiría evitar tratamientos inmunosupresores innecesarios con efectos secundarios indeseados, promover el trasplante renal de donante vivo, menor riesgo de recaída postrasplante renal, y dar la posibilidad de screening a los familiares y a las parejas de los pacientes.
Este estudio representa la primera descripción de las variantes de NPHS2 en un grupo de pacientes chilenos con SNCR<a 18 años de edad, así como también en un grupo de población general chilena en que se han estudiado variantes del gen NPHS2. Estos datos nos permiten conocer la epidemiología local del SNCR, mejorando el enfrentamiento clínico de la enfermedad de forma general, y de forma particular al permitir individualizar la causa de algunos de los pacientes que la presenten y ofrecer alternativas terapéuticas acorde a esta información.
FinanciamientoProyecto SOCHIPE 2012006. Proyecto Fondecyt 11090045.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
