


La interpretación inicial del proceso de coagulación mencionaba la presencia de 2 vías: la extrínseca, formada por el factor tisular (FT) y el factor VII, y la intrínseca, en la que participan los factores XII, XI, IX, VIII y V. Hoy en día este concepto ha cambiado y se acepta de forma categórica que el evento iniciador principal de la coagulación sanguínea es la exposición del FT. En este artículo revisamos los nuevos conceptos de la cascada de coagulación con los hallazgos de la tromboelastografía y los diferentes mecanismos que precipitan la coagulopatía asociada al trauma. Para tal fin se realizó una búsqueda sistemática en las principales bases de datos, como Medline, Embase y Lilacs, en el periodo comprendido entre 2000 y 2011. Se encontraron 114 artículos, de los cuales se tomaron 50 para realizar la revisión. Como hallazgo relevante se encontró que la tromboelastografía permite detectar con precisión el defecto subyacente en la cascada de coagulación, y de esta manera se ha convertido en una herramienta útil e indispensable para guiar el manejo de la coagulopatía asociada al trauma.
Initial interpretation of the process of coagulation refers to two pathways: (1) the extrinsic pathway, consisting of tissue factor (TF) and factor VII, and (2) the intrinsic pathway, in which factors XII, XI, IX, VIII and V are involved. Currently, this concept has changed and it is accepted that the main initiating event in blood coagulation is TF exposure. In this article, we review the new concepts of the coagulation cascade based on thromboelastography findings and the different mechanisms involved in trauma-associated coagulopathy. For this purpose, a systematic research was carried out using the main databases, including Medline, Embase and Lilacs between 2000 and 2011. One hundred and fourteen articles were found, and 50 were selected for the review. A relevant finding was that thromboelastography allows a precise detection of the underlying flaw in the coagulation cascade. Therefore, this procedure has become an essential tool and a guide for the management of trauma-associated coagulopathy.
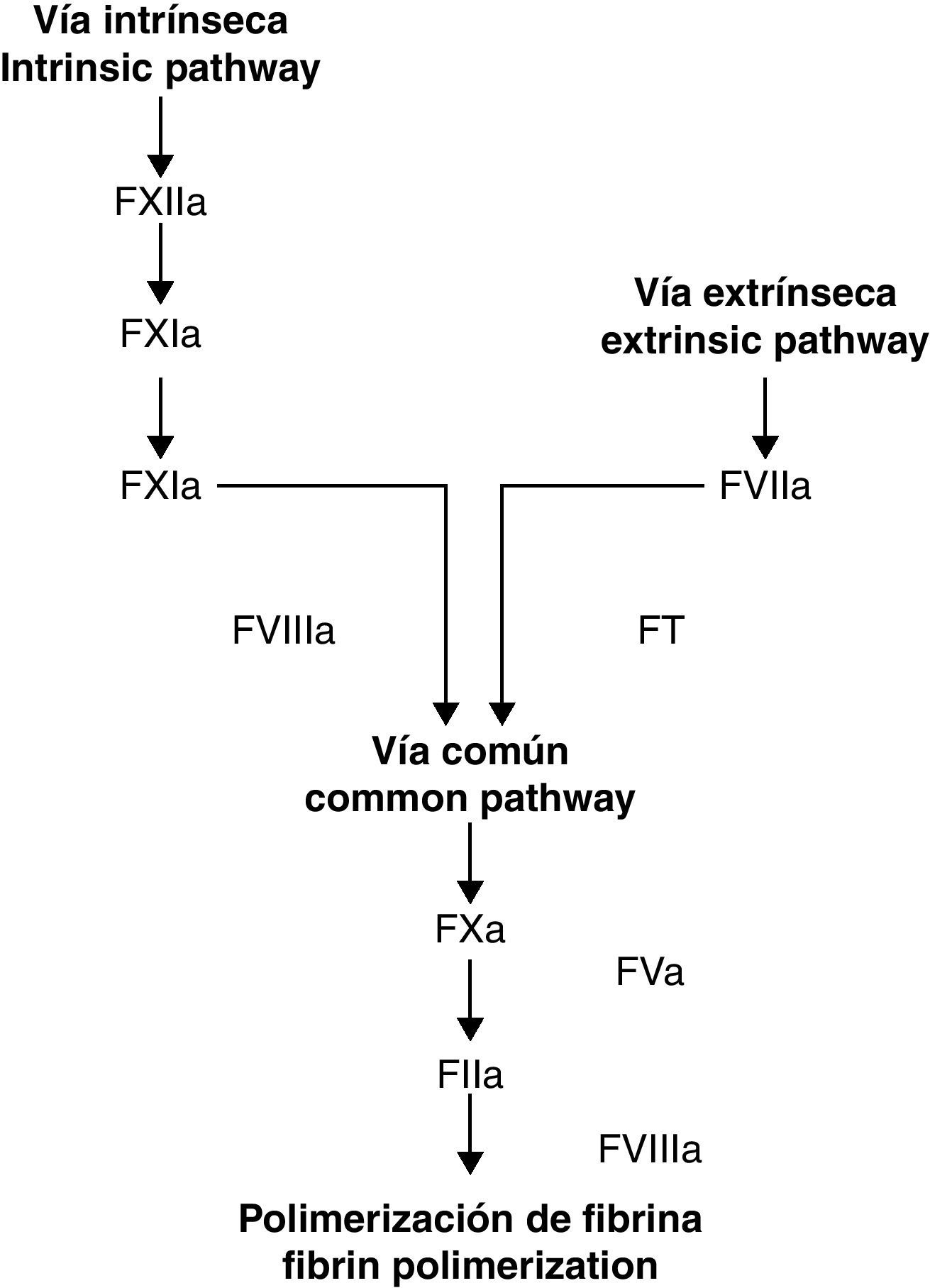
Durante décadas, se ha considerado que la cascada de coagulación tiene 2 puntos distintos de inicio, denominados vías extrínseca e intrínseca. Con el tiempo, no obstante, ha quedado claro que estas vías no funcionan en el organismo como sistemas paralelos e independientes1. El hecho demostrado de que el complejo factor tisular-factor VIIa (FT/FVIIa) de la vía extrínseca active factores en ambos sistemas indica que estas 2 vías están relacionadas2. Este descubrimiento, combinado con una comprensión cada vez mayor de la función de las plaquetas, ha dado lugar al modelo de coagulación celular3. A diferencia del antiguo modelo (vías intrínseca/extrínseca), el modelo celular incluye importantes reacciones entre las células directamente implicadas en la hemostasia (células receptoras del FT como el monocito y fibroblasto) y los factores de coagulación4.
MetodologíaSe realizó una exploración sistemática en las principales bases de datos, como Medline, Embase y Lilacs. Se seleccionaron las siguientes palabras clave: hemostasia, coagulación, tromboelastografía, coagulopatía y trauma. La búsqueda de información se llevó a cabo desde el año 2000 al 2011. Se encontraron un total de 114 artículos, cada uno se analizó de manera exhaustiva y finalmente se eligieron 50 documentos para efectuar la revisión.
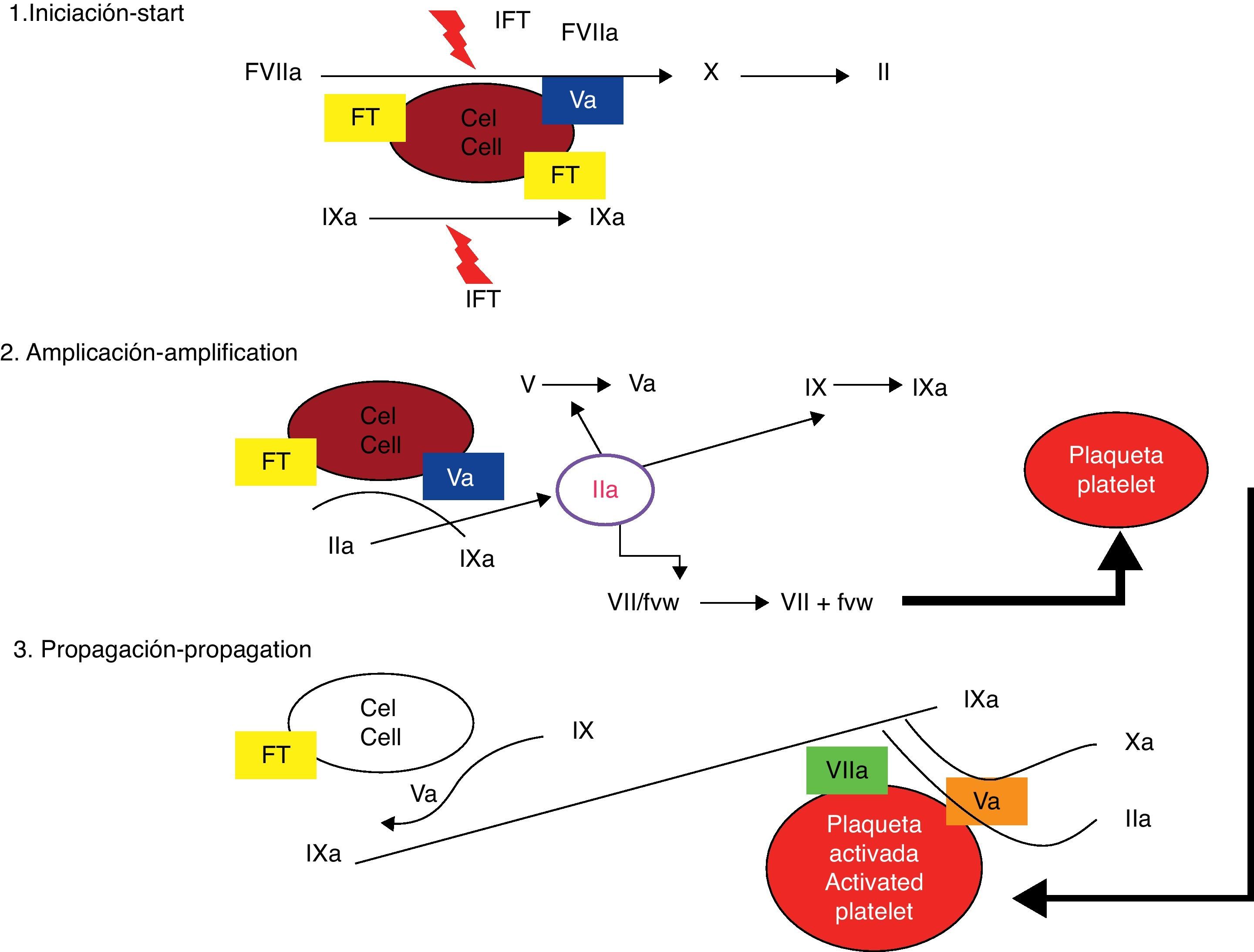
El modelo celularEl modelo celular identifica las membranas de las células receptoras del FT y las plaquetas como los lugares donde tiene lugar la activación de los factores de coagulación5. El modelo tradicional de la coagulación, propuesto hace 40 años, separaba las fases celular y humoral de la coagulación y considerada que el proceso de hemostasia se conseguía a través de la activación secuencial de enzimas efectoras en 2 vías independientes6. Recientemente se desarrolló un nuevo modelo que ha permitido un mejor entendimiento de cómo el sistema de hemostasia funciona in vivo (figs. 1 y 2). Esta nueva teoría, conocida como modelo celular de la coagulación, enfatiza la interacción entre los factores solubles y las superficies celulares y considera a las células como elementos esenciales capaces de dirigir el proceso hemostático7. El nuevo modelo resalta la importancia del complejo FT/FVIIa en la fase de activación del sistema y considera que la coagulación sucede en 3 fases que ocurren en distintas superficies celulares y de manera simultánea: iniciación, amplificación y propagación8,9.

Modelo clásico de la cascada de coagulación. Tomado de Jiménez et al.49.
 de la cascada de coagulación. Tomado de Jiménez et al.49.")
Modelo celular (moderno) de la cascada de coagulación. Tomado de Jiménez et al.49.
El complejo FT/FVIIa, de forma directa e indirecta a través del factor IX, activa inicialmente el factor X transformando pequeñas cantidades de protrombina en trombina, que son aún insuficientes para completar el proceso de formación de fibrina10. El FVII circula en la sangre predominantemente como molécula inactiva, y sus funciones, a las concentraciones fisiológicas, son virtualmente nulas en ausencia de su cofactor11,12. El FT no está en contacto con elementos de la sangre; la célula que alberga este receptor (fibroblasto, miocito, célula mononuclear, macrófago) se encuentra fuera del sistema vascular hasta que existe pérdida de la integridad del mismo13. La interacción entre el FT y FVIIa es el proceso fundamental en la iniciación de la coagulación; tal interacción incrementa la actividad del FVII en 1×107,14.
Fase de amplificaciónLa trombina así formada, junto con el calcio y los fosfolípidos ácidos que provienen de la plaqueta, participa activamente en un proceso de retroalimentación para la activación de los factores XI, IX, VIII y V, y de forma especial para acelerar la activación de la plaqueta15. Simultáneamente, por mecanismos quimiotácticos, los factores mencionados son atraídos a la superficie de las plaquetas, donde tienen lugar de forma muy rápida importantes procesos de activación y multiplicación16. La fase de amplificación es dependiente de la presencia de membranas plaquetarias activadas y de la interacción de estas con los factores de la coagulación, especialmente con las cantidades limitadas de trombina que se generan en la vecindad de la célula portadora del FT17. Las plaquetas se activan y degranulan, al tiempo que se adhieren y agregan formando un tapón en el vaso dañado. Una característica muy importante en la activación de las plaquetas es el cambio de polaridad de las cabezas negativas de los fosfolípidos para permitir su interacción con los factores de la coagulación18. Aunque es insuficiente para la formación de un coágulo, la pequeña cantidad de trombina producida por la vía VIIa/FT, durante la fase de iniciación, es esencial para amplificar el proceso19. La trombina es un ávido reclutador de plaquetas y retroalimenta de manera positiva al sistema al poseer la capacidad de activar los factores V, VIII y XI. La fase de amplificación también se caracteriza por la activación del sistema de retroalimentación negativa a través de los anticoagulantes naturales: TFPI (inhibidor de la activación del complejo FT/FVIIa), antitrombina y proteína C, cuya función es importante en regular los procesos procoagulantes20.
Fase de propagaciónLa amplificación del proceso por mecanismos de retroalimentación entre trombina y plaqueta y la activación de todos estos factores permiten activar grandes cantidades del factor X y formar el complejo protrombinasa para convertir la protrombina en trombina y, a expensas de esta, el fibrinógeno en fibrina21. El proceso final, siempre en la superficie de la plaqueta, se acelera para generar de forma explosiva grandes cantidades de trombina y fibrina. La fase de propagación presenta un cambio de locación de los procesos que llevan a la generación de la trombina, de la célula portadora de factor tisular a la plaqueta activada22. La presencia de fosfolípidos en la membrana plaquetaria activada permite el ensamblaje del complejo IXa/VIIIa y potencia sus acciones en 1×108. Grandes cantidades de trombina se producen durante esta fase, resultando en la escisión proteolítica del fibrinógeno y en la formación de monómeros de fibrina que se polimerizan para consolidar el inestable coágulo inicial de plaquetas en un firme coágulo organizado de fibrina. La trombina, a su vez, activa al factor XIII y al TAFI con efectos positivos adicionales en la estabilidad del coágulo y en la resistencia a los efectos de la plasmina23.
Papel de la plaquetaLa activación de la plaqueta altera la permeabilidad de la membrana y permite la entrada del calcio y la salida de sustancias quimiotácticas, que atraen a los factores de la coagulación a su superficie. Al mismo tiempo se liberan factor V y fosfolípidos ácidos, que aportan el complemento necesario para el proceso de la coagulación24.
En conclusión, la nueva cascada de coagulación presenta la formación de fibrina como resultado conjunto de 2 procesos: coagulación (representado por la trombina) y actividad de la plaqueta, que mutuamente se complementan.
FibrinólisisOtro proceso fundamental de la coagulación es la fibrinólisis, cuya función es eliminar los coágulos de fibrina durante el proceso de cicatrización, así como remover los coágulos intravasculares para impedir la trombosis25. El efector final del sistema es la plasmina, que degrada la fibrina en productos de degradación (PDF y dímero D). La plasmina es producida a partir de un precursor inactivo, el plasminógeno, por acción de 2 activadores del plasminógeno: activador tisular (t-PA) y activador tipo urocinasa (u-PA). La regulación de los activadores tiene lugar por la acción de inhibidores (PAI), de los que el más relevante es el PAI-1, mientras que la plasmina circulante es rápidamente inhibida por la α2-antiplasmina, lo que evita una fibrinólisis sistémica26. La fibrinólisis se inicia por el t-PA liberado desde el endotelio en respuesta a diversos estímulos (trombina, oclusión venosa, ejercicio físico, etc.). Una vez liberado, se une a la fibrina, donde activa el plasminógeno a plasmina que degrada la fibrina del coágulo27. La trombina puede activar otro inhibidor fibrinolítico, el TAFI, el cual elimina residuos de lisina de la fibrina, lo que impide la unión del plasminógeno y la ulterior degradación del coágulo28.
Monitoreo de la coagulación en el paciente con sangrado masivoExisten varias pruebas que valoran la coagulación, pero la tromboelastografía es la más útil para evaluar la coagulopatía del paciente traumatizado29. Esta prueba mide las propiedades viscoelásticas de la sangre de una forma dinámica y global, ya que integra las diferentes fases de la coagulación y la fibrinólisis, proporcionando una información dirigida a la detección de deficiencias del sistema hemostático30.
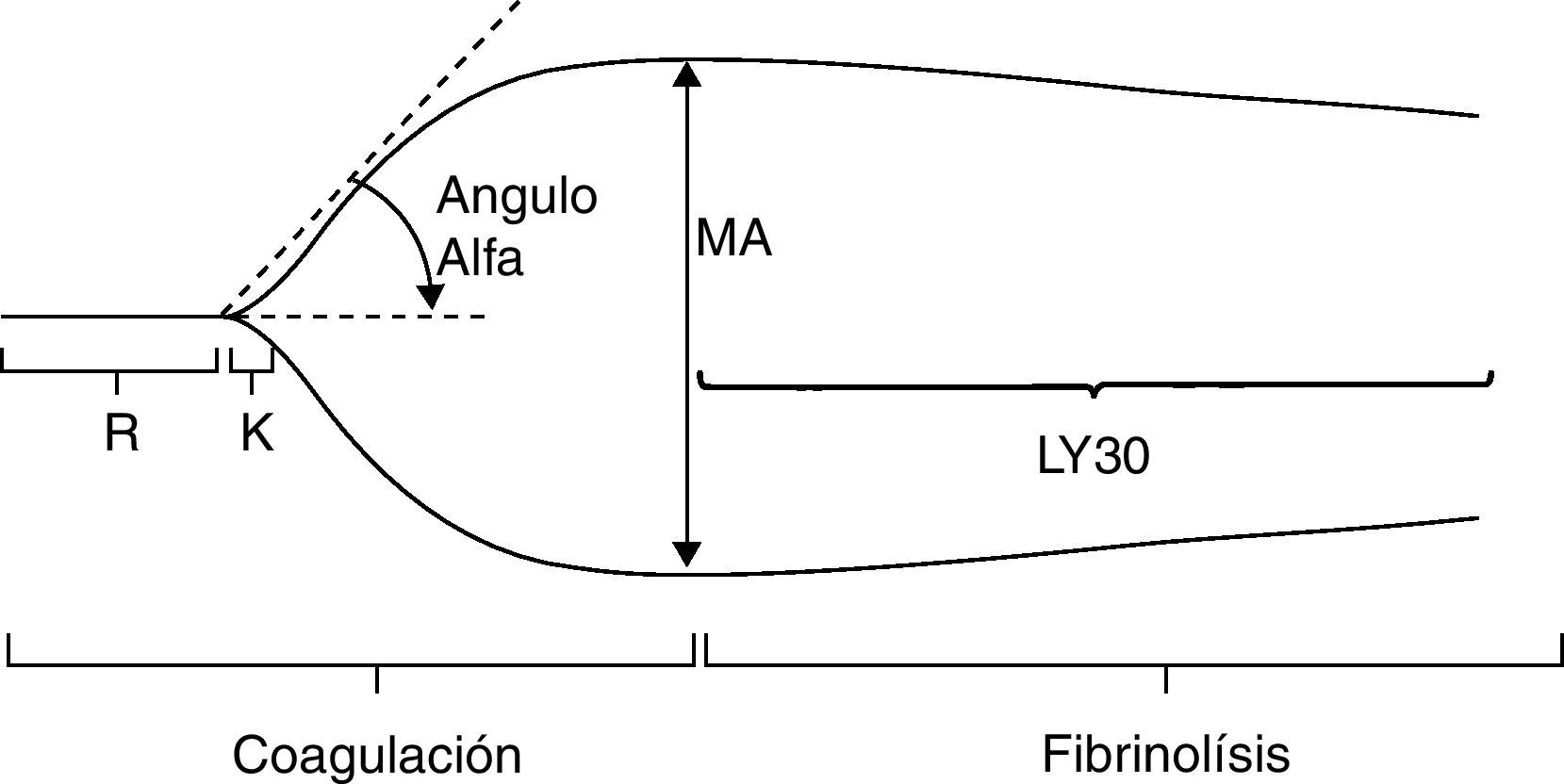
TromboelastografíaLa tromboelastografía (fig. 3) es la herramienta que permite medir las propiedades viscoelásticas de la sangre de una manera dinámica y global31. Fue desarrollada en Alemania en 1948 por Hartert, pero durante muchos años permaneció como una herramienta poco utilizada, y solo a mediados de los años ochenta el doctor Kang y colaboradores la retoman para el manejo de la coagulopatía durante el trasplante hepático y la cirugía cardíaca con circulación extracorpórea32. Desde ese momento su aceptación ha venido ganando terreno en diferentes campos de la medicina y ahora también se utiliza en anestesia obstétrica, anestesia del paciente traumatizado y en el paciente crítico que presenta coagulopatía22.

Tromboelastografía normal. Tomado de Gempeler et al.50.
La muestra para el tromboelastograma (TEG) es de sangre total, y debido a que esta es una prueba que con frecuencia se toma de pacientes en salas de cirugía y unidades de cuidados intensivos, puede obtenerse de los accesos invasivos como catéter central o línea arterial33. La muestra que se necesita es de 3cc, y se puede tomar en jeringa o en tubo citratado (tubo azul). El tubo citratado permite mucho más tiempo para su procesamiento (hasta 2h); sin embargo, se recomienda un tiempo estándar de 15min para iniciar la lectura. Esta prueba se realiza depositando 0,36cc de sangre en una copa, la cual requiere que se le ajuste la temperatura del paciente, y posteriormente se le introduce a esta muestra un pin, el cual se encuentra en un ángulo de 4°45”. Este pin es el encargado de traducir las propiedades físicas de la formación del coágulo, y mientras tanto la copa va a girar durante el tiempo en que la muestra cambia sus propiedades. El registro de estos cambios va a un dispositivo electrónico que posee un software encargado de esquematizar en una curva los resultados y expresar en números absolutos los parámetros a evaluar34.He aquí los valores convencionales de las diferentes fases de un TEG (fig. 3)35.
R: tiempo de reacción (minutos). Corresponde al intervalo entre el inicio de la coagulación hasta que el TEG tiene una amplitud de 2mm. Representa la velocidad de la generación de tromboplastina y refleja la función del sistema intrínseco, especialmente la actividad de los factores XII, XI y VIII. Se prolonga por deficiencias de factores de la coagulación y por consumo de anticoagulantes (warfarina, heparinas). Su acortamiento indica hipercoagulabilidad de cualquier origen. Su valor normal es de 4-8min.
R + K: tiempo de coagulación (minutos). Es el intervalo entre el inicio de la coagulación hasta que la amplitud del TEG es de 20mm. Mide la velocidad de formación de un coágulo de cierta solidez. Refleja la función del sistema intrínseco, las plaquetas y el fibrinógeno. En esta fase se alcanza el mayor aumento en la función plaquetaria y en la actividad de fibrinógeno, y se prolonga en caso de deficiencia de factores de coagulación o por consumo de antiagregantes plaquetarios. Se acorta cuando existe incremento en la función plaquetaria. Su duración es de 1-4min.
Ángulo alfa. Es el ángulo formado por el brazo de R y la pendiente de K, es la velocidad de formación de un coagulo sólido. Indica la calidad del fibrinógeno y de las plaquetas. Aumenta cuando existe hiperagregabilidad plaquetaria e hiperfibrinogemia y se reduce en casos de anticoagulantes y antiagregantes plaquetarios. Su valor normal es de 47-74grados.
MA: máxima amplitud (mm). Es la amplitud más grande que tiene el coágulo y es una función de la elasticidad del coágulo. Aumenta cuando mejora la calidad de las plaquetas, del fibrinógeno y del factor XIII. Evalúa la máxima medida del trombo y depende fundamentalmente de la interacción de la fibrina con las plaquetas. Su valor normal es de 55-73mm.
A60. Es la amplitud a los 60min de la MA (mm).
ILC: índice de lisis del coágulo (%), A60/MA. Es una medida en porcentaje que indica la proporción del coágulo que ha presentado fibrinólisis en un tiempo determinado, en este caso 30min. Su valor normal es del 0-8%, y cuando se encuentran valores mayores de 8% es necesario pensar en estados de hiperfibrinólisis tanto primaria como secundaria.
G. Parte de la máxima amplitud producto de la siguiente fórmula: 5.000 ma/(100 – ma), indica firmeza del coágulo, su valor se expresa en números absolutos y es muy sensible a cambios de máxima amplitud.
IC: índice de coagulación. Es un valor en números que pueden ser negativos y positivos. Su intervalo va desde –3 a +3; por debajo indica hipocoagulabilidad, y por encima, hipercoagulabilidad.
T: trombosis.
F: lisis del coágulo (minutos). Mide el intervalo desde la máxima amplitud (MA) hasta una amplitud 0 en el TEG y representa la actividad fibrinolítica.
Utilidad clínica de la tromboelastografíaEs un examen que puede realizarse en la cabecera del paciente, ofrece valiosa información del estado de coagulación y permite que la terapia transfusional pueda iniciarse más tempranamente y dirigirse hacia trastornos específicos como la disminución de factores de coagulación y/o la alteración plaquetaria (número y/o función)36.
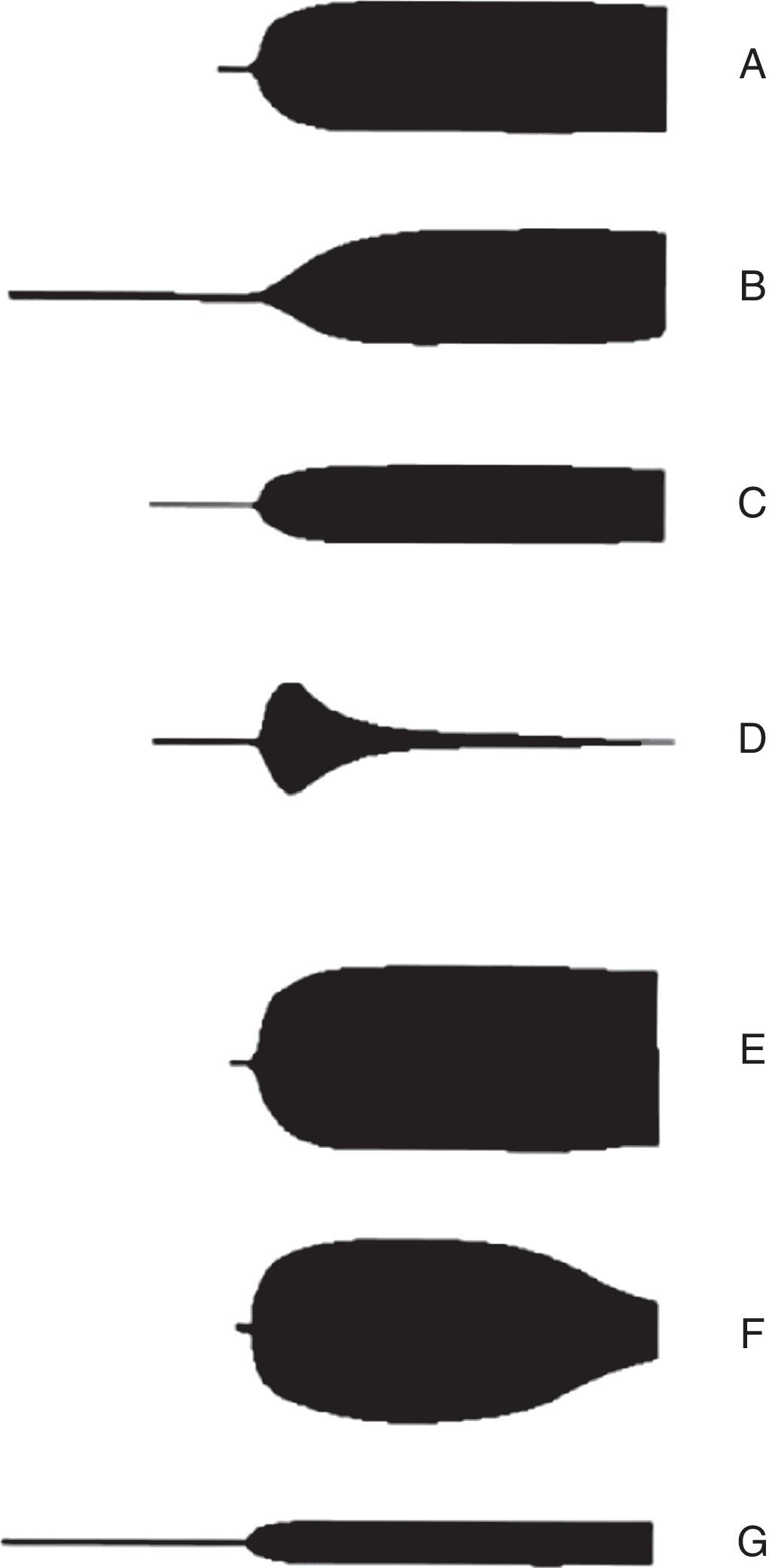
Dentro de las principales ventajas que ofrece, está el análisis in vitro de la relación entre los diferentes componentes de la coagulación, y de esta manera se puede observar la relación existente entre plaquetas, fibrinógeno y proteínas de la coagulación de forma integral. Frente a un paciente con sangrado que presenta cambios rápidos y complejos en el sistema hemostático, se hace indispensable información oportuna —ojalá en tiempo real y altamente confiable— para tomar decisiones efectivas37. La principal utilidad del TEG es que permite integrar las pruebas de coagulación convencionales con la función plaquetaria y así dar una idea más global de la fisiología de la hemostasia. Según los patrones de tromboelastografía (fig. 4), es posible identificar con precisión el defecto subyacente de la cascada de coagulación, lo que permite tener una idea clara de en qué fase se encuentra alterada y, de esta manera, facilita la decisión sobre el tratamiento que requiere el paciente38.
; C: amplitud máxima disminuida (trombocitopenia y bloqueadores de la función plaquetaria); D: fibrinólisis; E: hipercoagulabilidad; F: coagulación intravascular diseminada (CID); G: CID estadio tardío (hipocoagulabilidad). Tomado de Raffan-Sanabria et al.30.")
Patrones de la tromboelastografía. A: normal; B: prolongada (anticoagulación y deficiencia de factores); C: amplitud máxima disminuida (trombocitopenia y bloqueadores de la función plaquetaria); D: fibrinólisis; E: hipercoagulabilidad; F: coagulación intravascular diseminada (CID); G: CID estadio tardío (hipocoagulabilidad). Tomado de Raffan-Sanabria et al.30.
La coagulopatía del trauma es un proceso complejo, con daño tisular e hipoperfusión, que lleva a una serie de eventos que resultan en un estado hipocoagulable39. Aunque la hipotermia, la acidosis y la dilución de los factores de coagulación secundarios a la reanimación con cristaloides juegan un papel importante, la evidencia actual sugiere que la lesión tisular, la hipoperfusión, la fibrinólisis acelerada y los mecanismos inflamatorios también son factores cruciales en propiciar el desarrollo de la coagulopatía40,28.
Etiología de la coagulopatía en el paciente traumatizadoSon varias las causas que desencadenan el sangrado en un paciente traumatizado41:
- 1.
Coagulopatía dilucional. Dilución de los factores de coagulación y de las plaquetas tras la perfusión de grandes cantidades de volumen (coloides, cristaloides) en la reanimación inicial para mantener la volemia42.
- 2.
Hipotermia. Principal causa de coagulopatía en el shock traumático. Provoca disfunción plaquetaria severa y bloqueo enzimático de las reacciones fisiológicas de la coagulación43.
- 3.
Coagulopatía de consumo. La coagulación intravascular diseminada es un proceso patológico caracterizado por una masiva activación de la coagulación y por la reducción de la capacidad procoagulante44.
- 4.
Politransfusión. La transfusión abundante de sangre almacenada conlleva coagulopatía45. Siempre que el aporte de hemoderivados sea abundante, hay que tener en cuenta la denominada «lesión del banco de sangre», que está dada por:
- •
Dificultad en la entrega de oxígeno a los tejidos.
- •
Hipotermia.
- •
Intoxicación por citrato: disminución de calcio.
- •
Hipercaliemia.
- •
Acidosis.
- •
Hiperglucemia.
- •
- 5.
Fibrinólisis. Aunque es infrecuente, en las primeras fases del trauma puede aparecer un estado hiperfibrinolítico46.
- 6.
Acidosis. El valor del pH sanguíneo es el factor pronóstico más importante de coagulopatía, con una disminución en la actividad del FVIIa, del complejo FT/FVIIa y del complejo Xa/Va cuando el pH se aproximaba a valores de 7. La actividad enzimática de los factores de coagulación se encuentra disminuida hasta en el 90% a este pH47.
- 7.
Gravedad de la lesión. El traumatismo del tejido cerebral, las fracturas óseas y la presencia de líquido amniótico son situaciones clínicas en las que existe la posibilidad de embolismo, con los agravantes de que estos materiales son fuentes de tromboplastinas, contienen factor tisular y pueden causar coagulación intravascular diseminada, aumentando el consumo de los factores de coagulación48.
Los recientes avances relacionados con la fisiología de la cascada de coagulación han permitido entender con mayor exactitud la fisiopatología del sangrado en el paciente traumatizado. La utilización de la tromboelastografía ha llevado a que los médicos actuemos con mejores conocimientos en relación con los eventos que ocurren en el paciente y de esta manera ofrecer una terapia dirigida que esté encaminada a corregir el defecto hemostático de base. Todavía quedan muchas dudas por resolver sobre cuál sería el manejo ideal de la coagulopatía en trauma, pero la ciencia médica ha dado un gran avance en su manejo y ha abierto las puertas para que se generen nuevos conocimientos que permitan mejorar el manejo de esta patología.
FinanciaciónNinguna.
Conflictos de interesesLos autores declaran no tener ningún conflicto de intereses.