



Evaluar la equivalencia terapéutica de dos marcas comerciales de bisoprolol -hidroclorotiazida como terapia antihipertensiva.
MétodoEstudio prospectivo, doble ciego, doble falso, aleatorizado, de grupos paralelos, en el que se evaluó el efecto antihipertensivo de la combinación de bisoprolol-hidroclorotiazida 2,5-6,25 y 5-6,25mg (comprimidos BHL, formulación test) y bisoprolol-hidroclorotiazida 2,5-6,25 y 5-6,25mg tabletas (BHM, formulación de referencia), administrados en pacientes con hipertensión arterial.
Variables de efectividad: Presiones arteriales medidas mediante mediante esfigmomanómetro de mercurio al inicio y después del período placebo, a las 4 y 8 semanas del inicio del tratamiento; cambios horarios de la presión arterial durante 24 horas, mediante monitorización ambulatoria de la presión arterial.
ResultadosEl control de los valores de presión arterial se logró en ambas formulaciones, principalmente a partir de la cuarta semana de tratamiento. Los pacientes del grupo test ingresaron con presiones arteriales sistólicas más elevadas. Después del tratamiento no hubo diferencias entre los grupos, a ninguno de los tiempos. La relación V/P del grupo test fue 0,5-1. Los índices de suavidad de ambos fueron mayores a 1,75.
ConclusionesLa formulación test de la combinación de bisoprolol-hidroclotiazida demostró acción antihipertensiva similar al compararla con la formulación de referencia.
To evaluate the therapeutic equivalence of two commercial brands of bisoprolol –hydrochlorothiazide as antihypertensive therapy.
MethodA prospective, double blind, double placebo, randomised, parallel group study was conducted, in which the antihypertensive effect of the bisoprolol –hydrochlorothiazide 2.5 - 6.25mg and 5 - 6.25mg (tablets BHL, test formula) and bisoprolol -hydrochlorothiazide 2.5 - 6.25mg and 5 - 6.25mg tablets (BHM, reference formula), was compared by administering it to patients with arterial hypertension given to patients with arterial hypertension.
Effectivity variables: blood pressures measured using a mercury sphygmomanometer at the beginning and after the placebo period, at 4 weeks and 8 weeks from the start of the treatment; blood pressure hours change during 24hours using an ambulatory blood pressure monitoring device.
ResultsControl of the blood pressure values was achieved with both formulas, mainly from the fourth week of treatment. The patients of the test group were admitted with higher systolic blood pressures. After the treatment, there were no differences between the groups at any of the times. The V/P ratio of the test group was 0.5 – 1. The smoothness index in both groups was greater than 1.75 of fit of both was greater than 1.75.
ConclusionsThe test formula of the bisoprolol-hydrochlorothiazide demonstrated an antihypertensive action similar to that achieved with the reference formula.
En la actualidad la hipertensión arterial se destaca por ser una de las enfermedades que afecta en mayor medida a numerosas poblaciones, y posee tasas elevadas de complicaciones, así como de riesgo cardiovascular1. Según la Organización Mundial de la Salud (OMS), más de uno de cada cinco adultos poseen cifras de tensión arterial elevadas, que posteriormente presentan complicaciones como ataque cerebrovascular o cardiopatías2. En este sentido, en 2013 la literatura de la OMS, muestra datos estadísticos que afirman que aproximadamente mil millones de personas ya poseen un diagnóstico de hipertensión arterial, con una frecuencia de mortalidad anual promedio de nueve millones3.
Es por ello que cuando se trata esta enfermedad el objetivo es reducir al máximo la mortalidad producto de las complicaciones cardiacas y vasculares; de hecho, un tratamiento adecuado es capaz de disminuir la incidencia de ataque vascular encefálico en 35 a 40%, la de infarto agudo de miocardio en 20 a 25% y la de insuficiencia cardiaca en más del 50%4.
La evidencia epidemiológica acerca de los beneficios del tratamiento de la hipertensión arterial hace que se considere a los ensayos clínicos aleatorizados, como una opción que permite no solo demostrar la eficacia de los medicamentos, sino también comparar diferentes regímenes terapéuticos entre sí5. En la actualidad se dispone de una amplia gama de fármacos para tratar la hipertensión arterial, aunque algunos de ellos tienen efectos secundarios severos, lo cual los lleva, en la mayoría de los casos, a desuso, y por ende a que las investigaciones clínicas de la farmacoterapia de esta enfermedad continúen.
En este orden, con base en las fases iniciales de este tipo de investigaciones es preciso demostrar la eficacia de los fármacos frente a placebo; no obstante, cuando ya se cuenta con opciones terapéuticas eficaces, deben analizarse los nuevos fármacos frente a los tratamientos activos disponibles, lo que permite valorar su acción de forma comparada. En ambos casos estos estudios se basan en ensayos clínicos, en los que se busca demostrar que el fármaco experimental es mejor que el grupo control, sea placebo o grupo activo, mediante los llamados ensayos de superioridad, que pueden ser de eficacia o de seguridad6.
En el caso de estudios de eficacia, el fomento del uso de medicamentos para reducir el costo farmacéutico plantea la necesidad de verificar que cualquiera de los preparados investigados sea tan eficaz como el producto innovador o licenciado7. La falta de equivalencia terapéutica entre medicamentos con un mismo principio activo puede tener trascendencia médica en la eficacia del tratamiento terapéutico.
El medicamento original es aquel que contiene un principio activo innovador como consecuencia de un completo desarrollo en el campo de la farmacología preclínica y clínica. Los medicamentos que se contrastan pueden ser genéricos y contienen el mismo principio activo, en idéntica concentración e igual forma farmacéutica que el medicamento original e innovador, el cual es administrado con la misma posología y a las mismas indicaciones que el producto de referencia. Este tipo de medicamentos deben ser equivalentes, desde el punto de vista terapéutico, respecto al medicamento original y se comercializan luego del período de protección de patente del que goza el medicamento innovador u original8.
Es importante determinar la eficacia de los medicamentos con relación a los productos innovadores, ya que entre ellos existe diferencia económica, lo cual trasciende en la accesibilidad de estos en algunos usuarios9. En nuestro medio se hace necesario evaluar comparativamente los fármacos que se encuentran en el mercado nacional.
Para este estudio se tomará como tratamiento antihipertensivo al medicamento hemifumarato de bisoprolol, que es un antagonista beta 1 adrenérgico selectivo, desprovisto de actividad simpaticomimética intrínseca, y muy hidrosoluble en medios neutros y básicos. Este compuesto tiene un tiempo de semieliminación plasmática de 10 a 12 horas, una biodisponibilidad de 90% y efectos que perduran por 24 horas10.
Por otra parte, hoy en día existe la tendencia a la utilización de esquemas combinados de terapia antihipertensiva y se cuenta con numerosos estudios que demuestran que en la mayoría de los pacientes la monoterapia no es suficiente para alcanzar las metas terapéuticas de presión arterial y así disminuir el riesgo cardiovascular, por lo que las guías nacionales e internacionales vigentes recomiendan el uso de diversas combinaciones de fármacos antihipertensivos de acuerdo con sus comorbilidades o sus factores de riesgo acompañantes11. El Séptimo Informe del Comité Nacional Conjunto sobre Prevención, Detección, Evaluación y Tratamiento de la Hipertensión Arterial (JNC VII) publicado en 2003, recomienda iniciar el tratamiento farmacológico con cualquier agente de los diversos grupos de antihipertensivos y señala que preferentemente se combine con un diurético tiazídico en dosis baja12.
El objetivo de este estudio fue evaluar la equivalencia terapéutica de dos marcas comerciales de bisoprolol asociado a dosis bajas de un diurético tiazídico como terapia antihipertensiva, comparando la eficacia de ambos medicamentos.
Materiales y métodosSe trató de un estudio controlado, observacional, longitudinal, comparativo, doble-ciego, en el que los pacientes fueron distribuidos al azar, en dos grupos paralelos. El protocolo fue aprobado por el Comité de Ética de las instituciones en donde se realizó y todos los pacientes recibieron suficiente información y firmaron consentimiento escrito para participar en el mismo.
Se incluyeron pacientes de ambos sexos, adultos, con cifras de presión arterial diastólica (PAD) entre 90 a 99mm Hg y sistólica (PAS) de 140-179mm Hg, sin tratamiento previo o con tratamiento antihipertensivo, no controlados con monoterapia, y que requirieran una combinación de dos antihipertensivos de diferentes clases farmacológicas para el control óptimo de su hipertensión debido al fracaso terapéutico previo. Los pacientes con tratamiento antihipertensivo previo ingresaron después de un período de lavado de dos semanas, después de las cuales debían tener presión arterial diastólica mayor a 90 y presión arterial sistólica entre 140 y 180mm Hg.
En este estudio no se consideraron pacientes con evidencia de hipertensión arterial secundaria y diagnóstico previo de infarto del miocardio o de ataque cerebrovascular de cualquier tipo dentro de los seis meses antes del inicio del estudio. También se excluyeron pacientes con diagnóstico previo de asma o enfermedad pulmonar obstructiva crónica, arritmias cardíacas, insuficiencia cardíaca congestiva por encima del grado I NYHA13, niveles de creatinina sérica mayores a 2mg/dl, pacientes embarazadas o lactantes; con trastornos severos de la función hepática, diabetes mellitus mal controlada, historia de abuso de alcohol o drogas o pacientes con hipersensibilidad al bisoprolol o la hidroclorotiazida.
Durante el tiempo que duró la investigación no se permitió el uso de terapias con corticosteroides, anfetaminas, ciclosporina, eritropoyetina o combinaciones de medicamentos con oximetazolina, fenilefrina o pseudoefedrina, ni el uso concomitante de otros medicamentos antihipertensivos distintos a los utilizados en este estudio.
Antes de prescribir la combinación se hizo examen físico (presión arterial, pulso, peso, talla e índice de masa corporal) e historia médica (antecedentes personales y familiares, tratamiento y enfermedades asociadas), hematología completa, química sanguínea (glucosa, hemoglobina glucosilada o HbA1C, urea, creatinina, colesterol total y fraccionado, triglicéridos, sodio, cloro, potasio y transaminasas), así como electrocardiograma de control en la visita inicial. Lo anterior se hizo con el fin de retirar o incluir pacientes de acuerdo con los criterios antes señalados.
La presión arterial medida en el consultorio se tomó mediante un esfigmomanómetro de mercurio, en tres oportunidades separadas por 5 minutos, con el sujeto en posición sentado, con los pies apoyados en el piso y la espalda posada en la silla, con el aparato a la altura del corazón.
Se consideró la fase I y V de Korotkoff para determinar las presiones sistólica y diastólica respectivamente y se registró el valor promedio de las tres tomas para ambas presiones, de acuerdo con los lineamientos para el manejo de la hipertensión arterial del ESH/ESC 200714.
Los pacientes se distribuyeron al azar en cualquiera de los dos grupos de tratamiento.
- Bisoprolol-hidroclorotiazida formulación test 2,5-6,25mg, comprimidos (BHL). E.F. 34.763.
- Bisoprolol-hidroclorotiazida formulación de referencia 2,5-6,25mg, tabletas (BHM). E.F. 29.830.
A todos los pacientes incluidos en el estudio se les hizo monitorización ambulatoria de la presión arterial (MAPA) de 24 horas y se inició el tratamiento entre las 07:00 y las 09:00 a.m, durante 28 días consecutivos. Para el MAPA el período diurno se consideró desde las 06:00 a.m. hasta las 10:00 p.m, y las mediciones fueron hechas cada 15 minutos; entre tanto, el período nocturno fue desde la 10:01 p.m. hasta las 05:59 a.m. y las mediciones se programaron cada 20 minutos.
Si a las cuatro (04) semanas de tratamiento se habían logrado las metas (PAD ≤ 90mm Hg y PAS ≤ 140mm Hg en pacientes menores de 60 años o de PAD ≤ 90mm Hg y PAS ≤ 150mm Hg en pacientes mayores de 60 años)15 se hacía un nuevo MAPA de 24 horas, después de suministrada la última dosis y de esta manera el paciente finalizaba el estudio.
Cuando no se lograban las metas se administraba por cuatro semanas adicionales:
- Bisoprolol-hidroclorotiazida, formulación test 5-6,25mg, comprimidos (BHL). E.F. 34.764 o
- Bisoprolol-hidroclorotiazida, formulación de referencia 5-6,25mg, tabletas (BHM). 29.831.
Estos últimos pacientes recibieron tratamiento durante ocho semanas en total y se les hizo de igual manera un MAPA de 24 horas inmediatamente después de ingerir la última dosis de medicamento. Adicionalmente, a todos se les registró la presión arterial en el inicio o pretratamiento, a las 4, y a las 8 semanas del estudio.
Las variables principales de evaluación de la eficacia del tratamiento fueron los descensos en la presión arterial y los cambios de algunos parámetros en los MAPA de 24 horas realizados (presión periférica media y central media de 24 horas en la PAS y PAD).
También se consideraron los cambios en el MAPA en parámetros como el índice de aumentación o AIx 75 y la velocidad de onda de pulso (VOP). El primer parámetro se ajustó en función de la frecuencia cardíaca, equiparándose a un valor estándar de 75 latidos por minuto (lpm), mientras que la VOP se expresó en metros sobre segundo (m/s). Ambas son consideradas como parámetros mixtos de medición de la estructura y la función vascular, por lo que determinan fundamentalmente la rigidez arterial, en cuyo caso el sistema renina-angiotensina-aldosterona (SRAA) es uno de los principales mecanismos responsable de dichos cambios. Se consideró como valor de referencia una VOP menor a 9 m/s.
Así mismo, la valoración de la eficacia del fármaco a lo largo de las 24 horas se realizó mediante la evaluación de algunos índices numéricos que definieron el perfil farmacológico de un antihipertensivo, como el índice valle-pico o índice TP, que no es más que la relación entre la disminución de presión arterial al final del intervalo de la dosis (valle) y la máxima disminución conseguida después de administrar el fármaco (pico), así como el índice de alisamiento o smoothness index. Ambos indicadores se utilizaron para identificar si el antihipertensivo posee un perfil de reducción de la presión arterial adecuado durante el período de dosificación. Los valores normales deseables de estos parámetros fueron para el índice TP mayor a 0,5 o mayor a 50% y para el índice de alisamiento mayor a 116.
Análisis estadísticoTodos los datos relacionados con los pacientes fueron registrados en los respectivos formularios de evaluación clínica para el análisis estadístico. Se utilizó el programa estadístico SPSS versión 18. Las variables continuas se reportan como medias ± desviación estándar y las categóricas como porcentajes de casos.
Los datos que siguieron una distribución normal fueron evaluados estadísticamente mediante métodos paramétricos como la “t” de Student. Las variables medidas en una escala ordinal se analizaron por métodos no paramétricos, como el test de Wilcoxon. Las variables cualitativas se evaluaron mediante la prueba de chi-cuadrado o Woolf “G”. El nivel de significación se fijó en 0,05, con un intervalo de confianza del 95%.
ResultadosParticiparon un total de 80 pacientes, de los cuales sólo 62 (77,5%) arrojaron datos suficientes para realizar el análisis (38 grupo test vs. 24 grupo de referencia). El 58,8% de estos pacientes eran mujeres y 45,2% hombres. La edad, la PAS, la PAD y el pulso promedio de los pacientes totales estudiados fue 47,9 ± 8,9 años, 147,7 ± 11,3mm Hg, 98,2 ± 11,3mm Hg y 78,5 ± 11,0ppm respectivamente.
Dieciocho pacientes (22,5%) fueron excluidos del estudio por manifestar que no cumplieron con el tratamiento debido al olvido en la administración de la dosis o por causas ajenas al estudio no pudieron culminar las semanas de duración del estudio. El mayor porcentaje de abandono del estudio fue del grupo de referencia (40,0%). No hubo alteraciones en el electrocardiograma en los pacientes estudiados y 22 pacientes (57,9%) en el grupo test y 15 (62,5%) del grupo de referencia presentaron enfermedades asociadas, como sobrepeso, obesidad, diabetes mellitus controlada y dislipidemia. No se reportaron efectos adversos.
En cuanto al uso de una u otra combinación en el grupo test, 18 pacientes (52,7%) comenzaron y terminaron con la dosis de bisoprolol-hidroclorotiazida 2,5-6,25mg, y los restantes 20 (47,3%) comenzaron con bisoprolol-hidroclorotiazida 2,5-6,25mg y terminaron con la dosis de bisoprolol-hidroclorotiazida 5,0-6,25mg; mientras que enel grupo de referencia 8 pacientes (33,3%) inició y terminó con la dosis más baja y 16 pacientes (66,7%) terminó cuatro semanas después con la dosificación más alta.
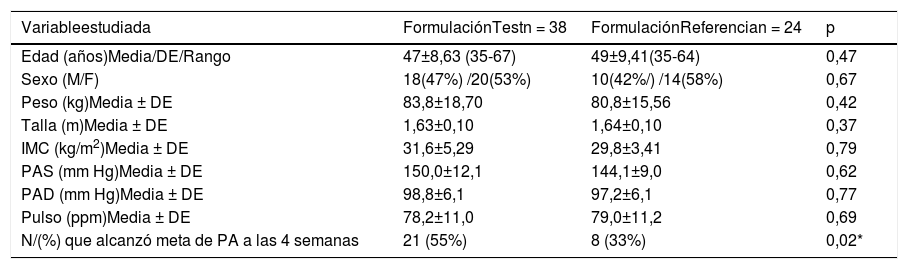
Las características de los grupos estudiados se describen en la tabla 1. No se observan diferencias significativas en cuanto a edad, sexo, peso, talla, índice de masa corporal, PAS, PAD y pulso. Sólo 29 (46,8%) de los pacientes totales estudiados alcanzaron la meta de presión arterial a las 4 semanas, de los cuales 21 (55,3%) pertenecían al grupo test y 8 (33,3%) al grupo de referencia, con diferencias significativas al comparar ambos grupos (p<0,05).
Descripción de los pacientes estudiados
| Variableestudiada | FormulaciónTestn = 38 | FormulaciónReferencian = 24 | p |
|---|---|---|---|
| Edad (años)Media/DE/Rango | 47±8,63 (35-67) | 49±9,41(35-64) | 0,47 |
| Sexo (M/F) | 18(47%) /20(53%) | 10(42%/) /14(58%) | 0,67 |
| Peso (kg)Media ± DE | 83,8±18,70 | 80,8±15,56 | 0,42 |
| Talla (m)Media ± DE | 1,63±0,10 | 1,64±0,10 | 0,37 |
| IMC (kg/m2)Media ± DE | 31,6±5,29 | 29,8±3,41 | 0,79 |
| PAS (mm Hg)Media ± DE | 150,0±12,1 | 144,1±9,0 | 0,62 |
| PAD (mm Hg)Media ± DE | 98,8±6,1 | 97,2±6,1 | 0,77 |
| Pulso (ppm)Media ± DE | 78,2±11,0 | 79,0±11,2 | 0,69 |
| N/(%) que alcanzó meta de PA a las 4 semanas | 21 (55%) | 8 (33%) | 0,02* |
*p<0,05.
kg: kilogramo; m: metro; IMC: índice de masa corporal; PAS: presión arterial sistólica; PAD: presión arterial diastólica; mm Hg: milímetros de mercurio; ppm: pulsaciones por minuto.
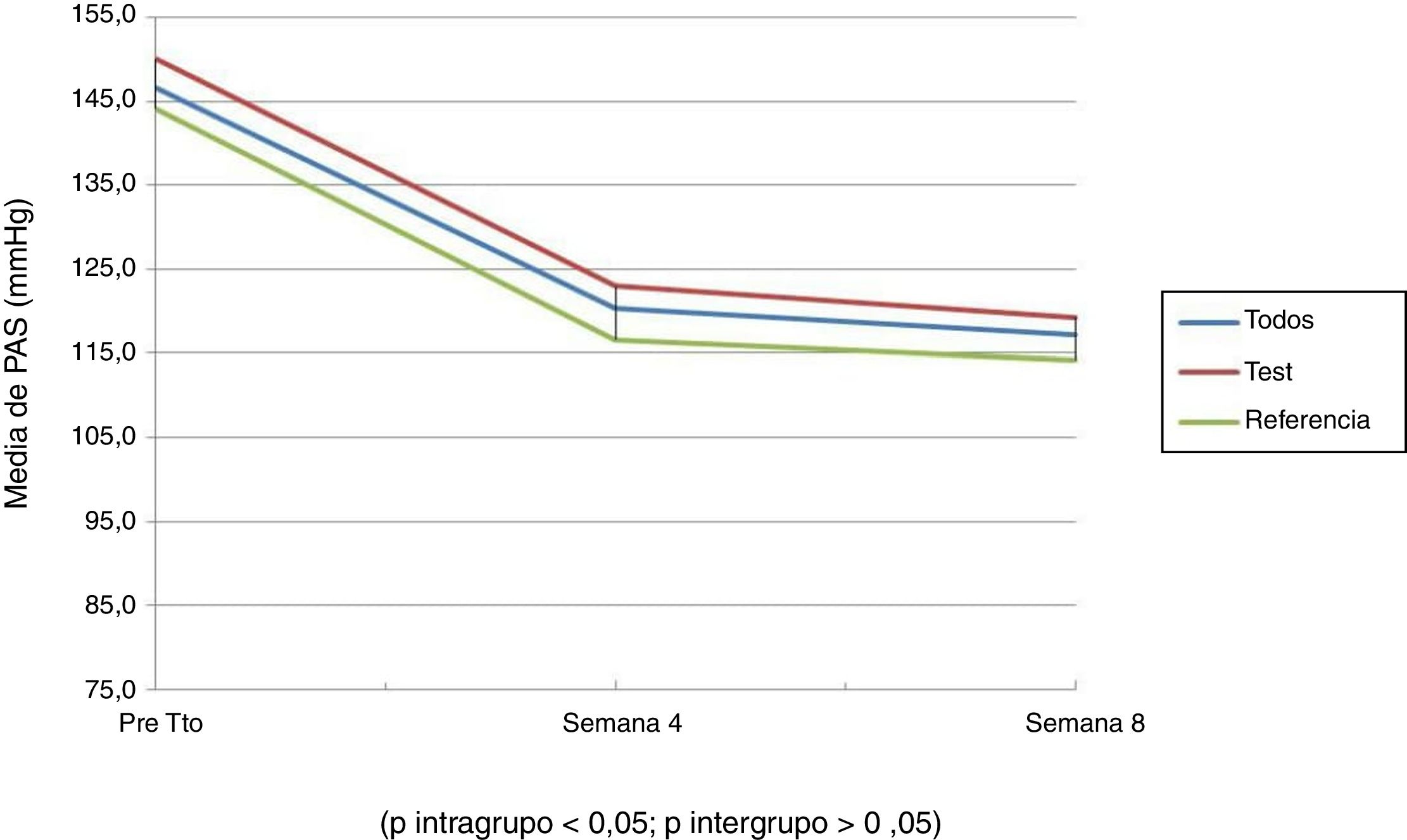
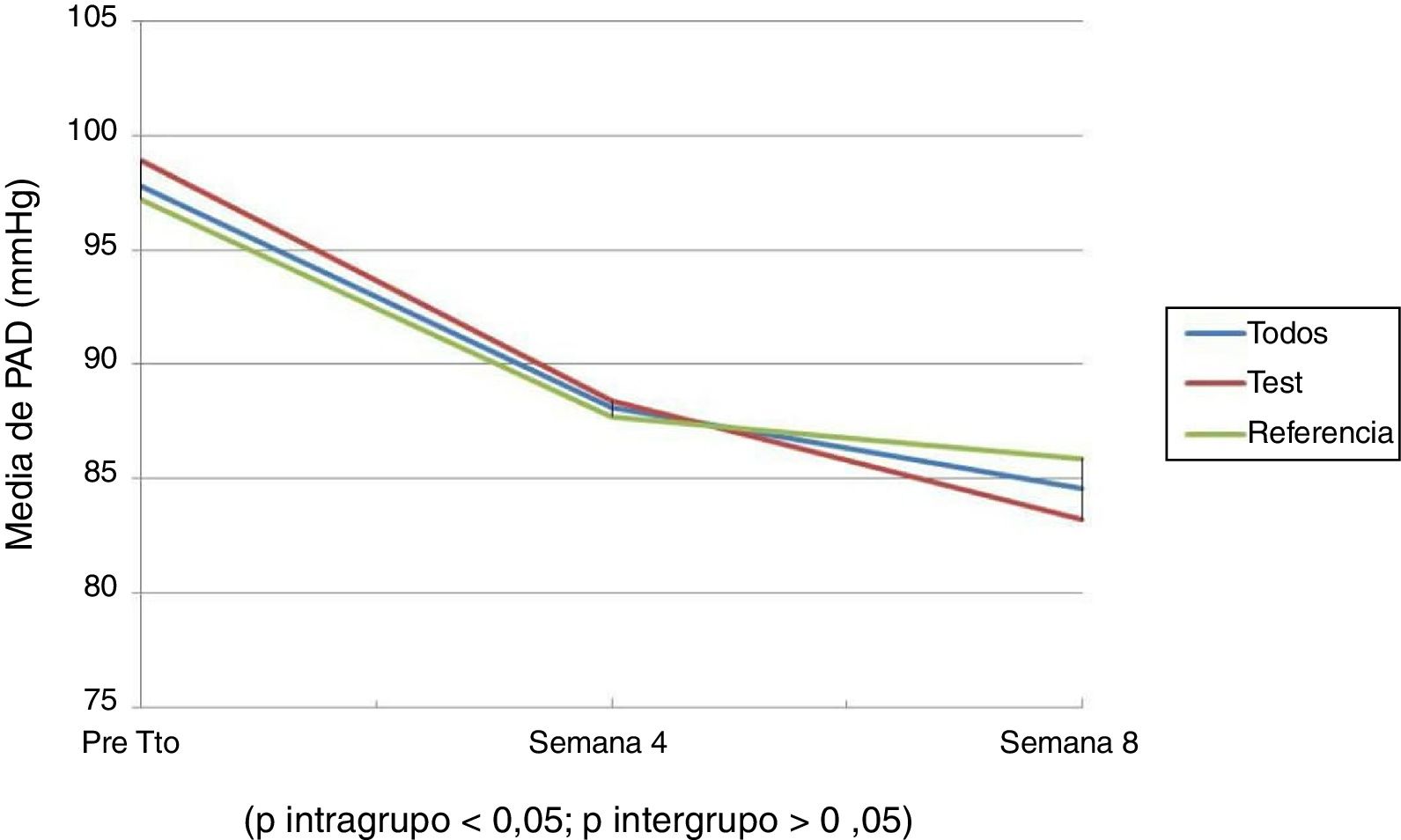
Las figuras 1 y 2 muestran la evolución de la PAS y la PAD; se puede observar que hubo un descenso en el promedio de la PAS y la PAD, en la semana 4 y 8 del tratamiento, que al compararlas (diferencias intragrupo) fueron estadísticamente significativas. No se observó diferencias significativas al comparar ambos grupos (diferencias intergrupo), es decir entre los pacientes que recibieron la formulación test y de referencia. En ambos grupos estudiados el descenso promedio de la PA siguió el mismo comportamiento.


Cuando se compararon las presiones periférica media y central de 24 horas para PAD y PAS en la formulación test en la monitorización de presión arterial (MAPA) antes y después del tratamiento se observaron diferencias significativas. También, hubo una ligera reducción del índice AIx75 y de la VOP, pero sin modificación significativa de estos parámetros. Valores similares se registraron para la formulación de referencia.
Al estudiar las variables de eficiencia del tratamiento evaluadas en el MAPA realizado una vez culminado el tratamiento (tabla 2) en ambos grupos, no se observaron diferencias significativas en los parámetros. En ambos grupos estudiados se reportaron valores promedio similares.
Variables de eficiencia al tratamiento evaluadas con la monitorización ambulatoria de la presión arterial (MAPA) comparando la formulación test y de referencia
| Variable estudiada | FormulaciónTestn = 38 | Formulación Referencian = 24 | p |
|---|---|---|---|
| PAS periférica media 24 hMedia ± DE | 122,67±9,08 | 117,50±10,69 | 0,14 |
| PAS central media 24 hMedia ± DE | 116,11±8,57 | 109,41±8,65 | 0,20 |
| PAD periférica media 24 hMedia ± DE | 81,80±7,71 | 77,21±7,9 | 0,09 |
| PAD central media 24 hMedia ± DE | 83,05±8.77 | 77,55±7,12 | 0,10 |
PAS: presión arterial sistólica; PAD: presión arterial sistólica.
De igual manera, al medir el índice valle-pico o índice TP, este alcanzó valores de 0,6 (60% de mantenimiento del efecto antihipertensivo), y el índice de alisamiento o smoothness index obtuvo valores de 1,75.
DiscusiónUna de las mayores expectativas de los investigadores y de los clínicos, cuando se estudia un fármaco nuevo, es encontrar evidencias de superioridad del nuevo medicamento respecto al tratamiento estándar que se esté utilizando, de manera que el nuevo producto permita un avance en la terapia de los pacientes17. Sin embargo, también es cada vez más frecuente que los estudios se enfoquen en demostrar que el fármaco que se investiga sea igual de eficaz que otro considerado como de referencia.
Este tipo de investigaciones relacionadas con el establecimiento de equivalencia terapéutica han tomado en la actualidad un gran impulso y han adquirido importancia desde el punto de vista clínico, así como de gestión18. En cuanto a clínica, se puede establecer si es necesario disponer de varios equivalentes terapéuticos en la guía farmacoterapéutica, considerando así segundas opciones de tratamiento, o bien disponer sólo de uno de ellos. Desde el punto de vista de la gestión, con estos estudios se demuestra si conviene disponer de un solo equivalente o bien poder definirlos como medicamentos homólogos, lo que permite la inclusión de varios medicamentos, cuyos principios activos se definen como equivalentes terapéuticos y que se utilizarán indistintamente, en función de los costos o de la disponibilidad en el mercado.
Laguna et al. afirman que en muchos países antes de comercializar los medicamentos se exige garantizar su calidad, seguridad y eficacia. Los medicamentos llamados multifuentes o genéricos deberán demostrar seguridad y eficacia a través de estudios de equivalencia terapéutica contrastando con el medicamento original, con el fin de garantizar su intercambiabilidad19.
En este estudio ambos grupos fueron comparables en cuanto a edad, peso, talla e índice de masa corporal; no obstante, en los dos prevaleció el sexo femenino. En estudios similares llevados a cabo en Brasil20, también hubo mayor cantidad de mujeres, lo cual obedece a una mayor adhesión de la mujer al tratamiento ya que los hombres, con promedio de edades para ese estudio, aún trabajan activamente, hecho que no permite una mayor participación.
Algunos factores de riesgo asociados que se encontraron en los sujetos estudiados, merecen comentario. Es bien sabido que, de acuerdo con el estudio de Framingham, entre el 60 y 70% de los hipertensos se atribuye directamente al exceso de adiposidad21, mientras que la hipertensión arterial es una comorbilidad que afecta el 20 al 60% de la población con diabetes mellitus, contribuyendo en el desarrollo y la progresión de las complicaciones crónicas de la diabetes22.
Se hace necesario discutir que aunque cuando hubo diferencias significativas al comparar el porcentaje de pacientes que alcanzaron la meta de PA, este hallazgo pudo deberse a que uno de los grupos estudiados tuvo menor número de participantes, debido a abandono del estudio o bien por no concluir las semanas de estudio.
Por otra parte, esta investigación demostró efectividad en la reducción de la PA tal como muestran las figuras 1 y 2, siendo ambos fármacos de comportamiento similar. De igual manera, no se reportaron efectos adversos y hubo buena tolerabilidad. Otros estudios similares realizados al azar y doble ciego23,24 han demostrado también la tolerabilidad, seguridad y eficacia del uso de bisoprolol combinado con hidroclorotiazida en la reducción de la PA, cuyo efecto se mantuvo durante 24 horas y con mínimos efectos adversos. Estudios en este país realizados por Fuenmayor y Gómez25, indican que la combinación bisoprolol/hidroclorotiazida fue efectiva en la disminución de la PA desde la primera semana del tratamiento.
En consecuencia, estos resultados apoyan el concepto que la combinación a bajas dosis de un antagonista de los receptores adrenérgicos y un diurético actúa a través de mecanismos diferentes para reducir la PA, produciéndose así un efecto adicional que minimiza, a la vez, los efectos secundarios relacionados con las dosis y los efectos metabólicos, que se evidencian cuando se utilizan altas dosis de estos medicamentos26.
Los betabloqueantes y los diuréticos tiazídicos son fármacos considerados de primera línea en la reducción de la PA, que utilizan diferentes mecanismos. Los primeros reducen el gasto cardiaco al regular la frecuencia cardiaca, la PAS y la contractilidad ventricular; y los segundos, disminuyen las resistencias vasculares periféricas y la sensibilidad al sodio, generando vasodilatación periférica entre otros mecanismos. Ambos fármacos tienen actividad supresiva sobre la liberación de renina, por lo que la combinación es ampliamente considerada como muy potente27, tal como se corrobora en el presente estudio.
Las concentraciones involucradas en este estudio para cada fármaco (bisopropol-hidroclorotiazida) han demostrado eficacia antihipertensiva, conforme lo muestran las variables de eficacia estudiadas (tabla 2) y los índices que definen el perfil farmacológico de un antihipertensivo (índice valle-pico y el índice de descenso suave). Una de las principales indicaciones clínicas, donde esta última monitorización demuestra su utilidad, es en la evaluación de la eficacia antihipertensiva durante un periodo de 24 horas, lo cual facilita una prescripción más racional en lugar de basar la decisión en una o varias mediciones de consultorio, que sólo representan una pequeña parte del ciclo diurno de la tensión arterial28.
En contraste, estudios previos realizados por Neutel29 demuestran resultados similares, de modo que es claro suponer que en la práctica médica diaria administrar bisoprolol-hidroclorotiazida de manera conjunta tiene efecto sinérgico considerablemente bueno para alcanzar el control en pacientes con HTA, sin efectos secundarios relevantes.
Si bien el diseño del estudio no permitió tener mayor número de participantes, los resultados de la evaluación de la equivalencia terapéutica utilizando el método estadístico de la mínima diferencia significativa (LSD) fueron semejantes al evaluar ambos grupos, así que estos hallazgos resultan alentadores en la búsqueda de combinaciones farmacológicas eficaces como segunda opción terapéutica en pacientes con hipertensión arterial.
Se concluye que la formulación test de la combinación de bisoprolol/hidroclorotiazida demostró poseer una acción antihipertensiva significativa en el manejo de los pacientes con diagnóstico de hipertensión arterial al compararse con la formulación de referencia. El control de los valores de presión arterial se logró al igual en ambas formulaciones, principalmente a partir de la cuarta semana de tratamiento.
FinanciaciónEl estudio fue financiado por Laboratorios Leti, S.A.V, Guarenas, Venezuela.
Conflicto de interesesNinguno.