


La distrofia miotónica es una enfermedad hereditaria del sistema neuromuscular con afección cardiaca, se presenta fundamentalmente con alteraciones del ritmo y conducción auriculoventricular y rara vez insuficiencia cardiaca. Se expone el caso de un paciente de 37años de edad que ingresa por falla cardiaca aguda y flutter auricular asociado a debilidad muscular progresiva de larga data en quien se realiza diagnóstico de distrofia miotónica tipo1 luego de estudios complementarios. Se presenta una corta revisión de la literatura acerca de esta enfermedad y sus manifestaciones cardiacas.
Myotonic dystrophy is a hereditary disease of the neuromuscular system with cardiac impairment, mainly showing rhythm disturbances and atrioventricular conduction defects, and rarely heart failure. We report the case of a 37 year-old patient who was admitted for acute heart failure and atrial flutter associated with long standing progressive muscle weakness. A diagnosis of myotonic dystrophy type1 was made after complementary studies. A short review of the literature about this pathology and the cardiac manifestations is presented.
Paciente masculino de 37años de edad quien ingresa por cuadro clínico de 2meses de evolución consistente en disnea de esfuerzo progresiva, ortopnea y episodios de disnea paroxística nocturna. Sin antecedentes de cardiopatía. El paciente refiere que desde hace aproximadamente 8años presenta pérdida progresiva de la fuerza y movilidad de miembros superiores sin haber recibido evaluación médica; antecedente familiar de 2 primos con distrofia muscular no especificada y padre con falla cardiaca de etiología no clara que falleció a los 58años de edad.
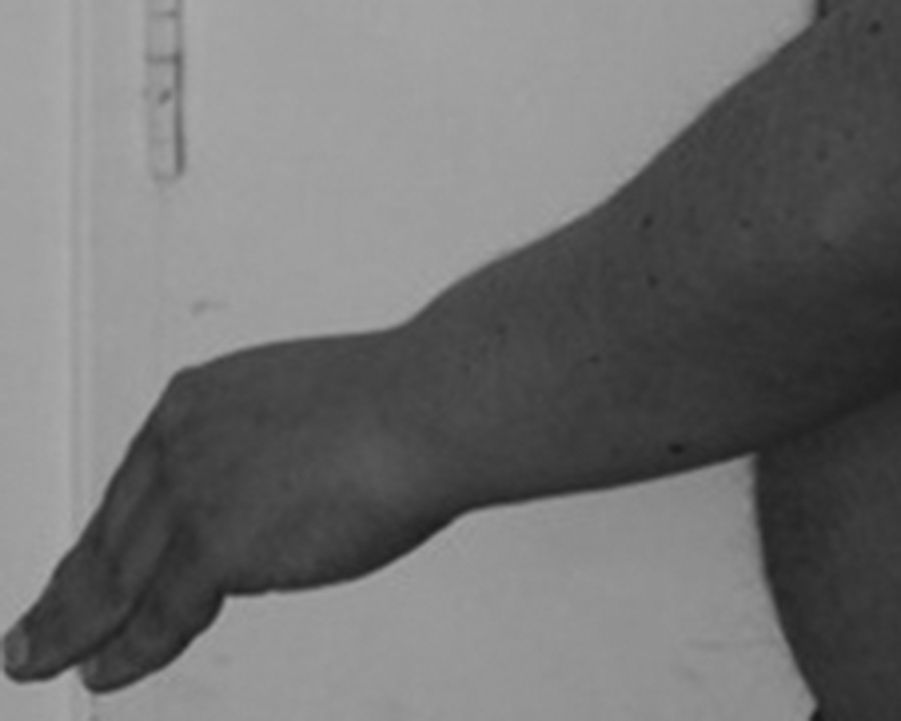
Al examen físico se encuentra presión arterial de 110/80mmHg, FC 110 latidos/min, afebril, fascies miopática con debilidad facial, atrofia de los músculos masticadores (fosas temporales con atrofia) y calvicie. En la auscultación cardiaca se encontraron ruidos cardiacos rítmicos, con soplo holosistólico mitraliii/vi, ingurgitación yugular de 10cm a 45° y crépitos bilaterales hasta el tercio medio de ambos campos pulmonares. En las extremidades se encuentra deformidad en las manos con actitud «distónica» (flexión en las articulaciones metacarpofalángicas e hiperextensión en las interfalángicas) (fig. 1), con respuesta miotónica a la percusión de los músculos intrínsecos de las manos; atrofia muscular generalizada, predominio en la cintura escapular y las piernas con fuerza muscular disminuida en los miembros superiores: proximal 2/5, distal 3/5, y los miembros inferiores: proximal 4/5, distal 5/5. Además, retracciones articulares en el codo, el tobillo y la columna cervical. Es llamativa la presencia de miotonía en la lengua y los músculos de las extremidades, con contracción sostenida y dificultad para la relajación muscular ante la percusión o al pedirle que apriete los puños y que los abra en forma rápida.

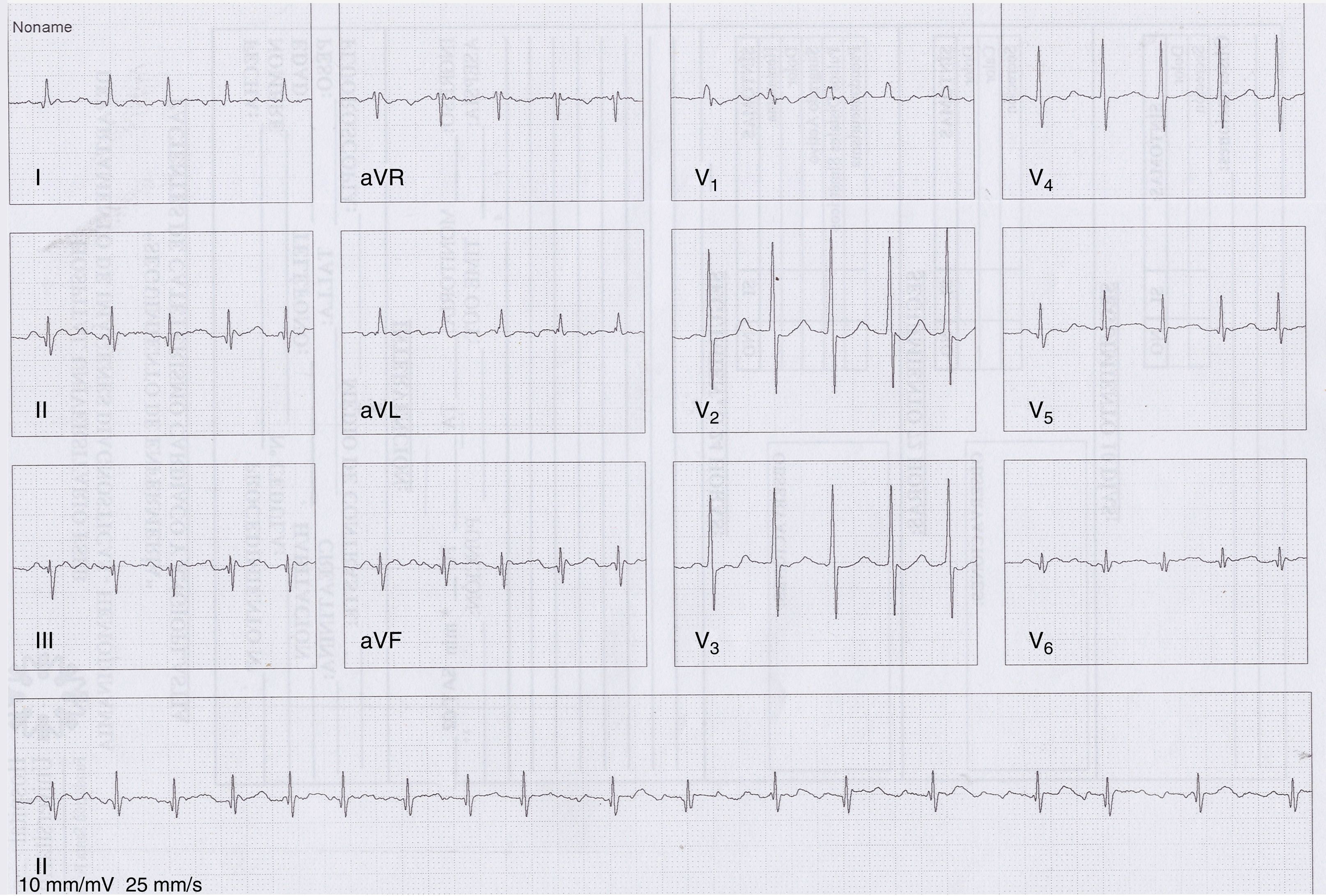
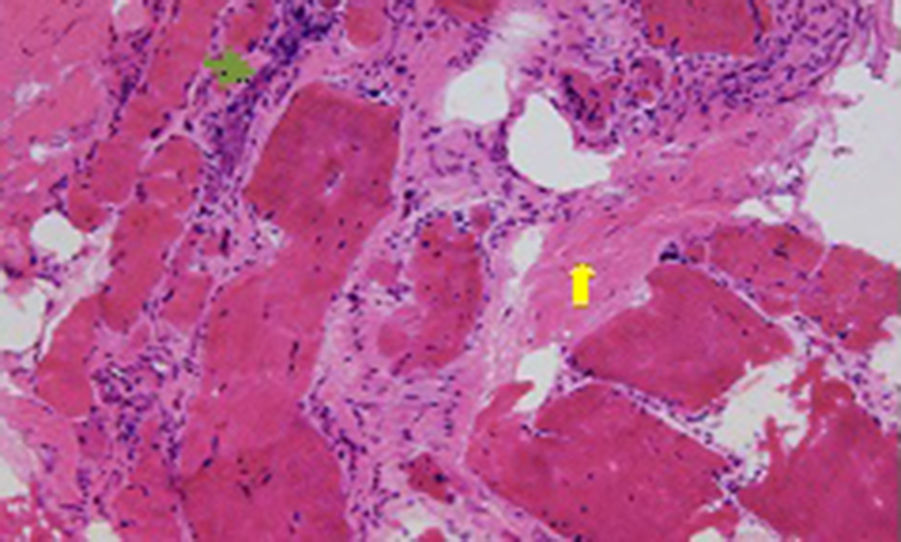
En la radiografía de tórax se aprecia aumento del índice cardiotorácico, redistribución de flujo vascular y patrón intersticial bilateral. El electrocardiograma muestra flutter auricular (fig. 2). En el ecocardiograma se aprecia un ventrículo izquierdo dilatado con hipocinesia generalizada, función ventricular deprimida (fracción de eyección del ventrículo izquierdo de 25%) y presencia de trombo intracavitario. El hemograma y las pruebas de coagulación son normales, al igual que la glucemia, la TSH y los electrólitos. Las serologías para virus de hepatitisB y C, Chagas y VIH fueron negativas. Las pruebas de neuroconducción son normales. En la electromiografía se evidencia una membrana muscular inestable con actividad de inserción aumentada con abundante actividad de potenciales agudos positivos y fibrilares, descargas miotónicas de frecuencia variable y un patrón miopático en músculos proximales y distales con unidades motoras polifásicas de baja amplitud, llenamiento temprano del patrón de interferencia con esfuerzo medio y salvas de potenciales miotónicos de frecuencia variable, espontáneas y provocadas por desplazamiento del electrodo o por percusión sobre el músculo. La biopsia muscular muestra un músculo marcadamente comprometido por infiltración por tejido adiposo, con mayor afectación de algunos fascículos que otros. Las fibras que lo componen muestran marcada variación en su tamaño, evidenciándose pocas fibras con cambios degenerativos o regenerativos. El cambio más prominente se trata de la internalización de los núcleos, que se encuentra presente en un 68% de las fibras, y en algunas la cantidad de núcleos es mucho más numerosa comparado con otras. También, se observaron algunas fibras en extremo atróficas formando sacos de núcleos. El colágeno endomisial se encuentra aumentado en forma severa. Se encuentran cambios degenerativos de las fibras (fig. 3). A nivel ultraestructural no fueron identificados cambios en la organización de las miofibrillas, anomalías de la heterocromatina y su fijación a la membrana nuclear, inclusiones nucleares, a pesar de su internalización, ni alteraciones mitocondriales (fig. 4).


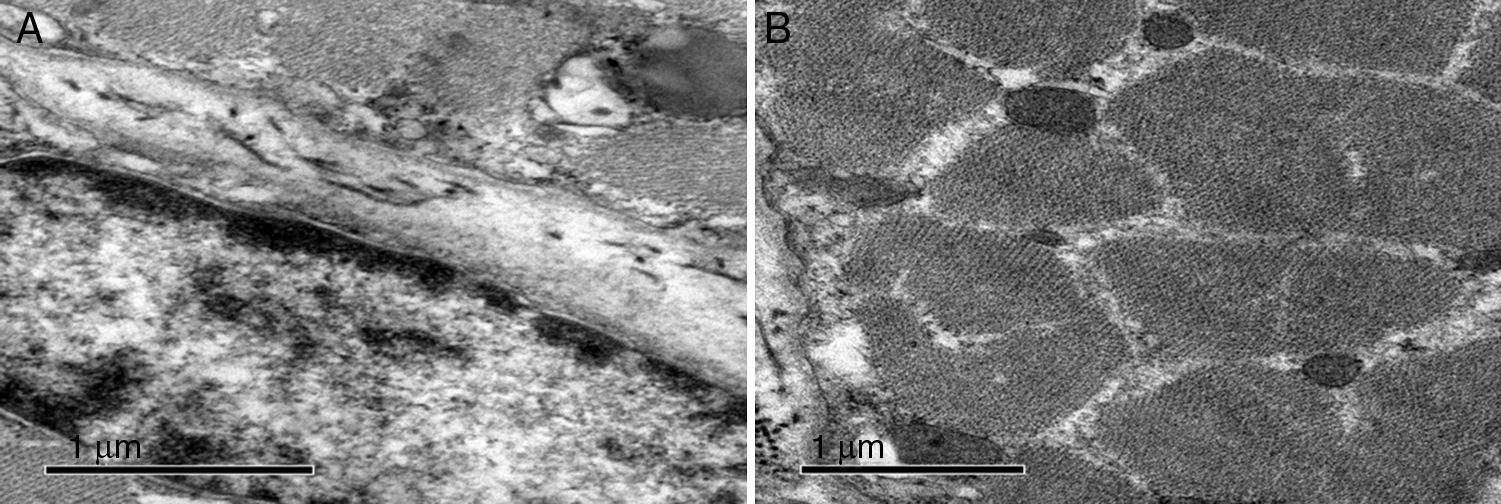
Se observa un núcleo en localización normal, en donde la heterocromatina se encuentra en íntimo contacto con la membrana nuclear. No hay defectos de la membrana. B)Corte transversal de las miofibrillas y su relación normal de las mitocondrias. Estas últimas no muestran alteraciones de sus crestas ni tienen anomalías de su tamaño o inclusiones. En las miofibrilas se encuentran intercalados los filamentos gruesos de la miosina y delgados de la actina en forma usual.")
Estudio de ultraestructura. A)Se observa un núcleo en localización normal, en donde la heterocromatina se encuentra en íntimo contacto con la membrana nuclear. No hay defectos de la membrana. B)Corte transversal de las miofibrillas y su relación normal de las mitocondrias. Estas últimas no muestran alteraciones de sus crestas ni tienen anomalías de su tamaño o inclusiones. En las miofibrilas se encuentran intercalados los filamentos gruesos de la miosina y delgados de la actina en forma usual.
Las distrofias musculares son trastornos hereditarios que abarcan un espectro amplio de afectación muscular progresiva como consecuencia de distintas alteraciones en genes codificantes de las proteínas de la membrana celular, el citoesqueleto y el núcleo de los miocitos. El compromiso cardiaco de estas enfermedades no es inusual.
Las manifestaciones cardiacas de estas enfermedades pueden variar dependiendo de cada tipo1. Las distrofias musculares de Duchene y Becker son trastornos genéticos recesivos ligados al cromosomaX, en las cuales se presenta una mutación en el gen distrofina que codifica una proteína del mismo nombre. La distrofina y las glucoproteínas asociadas a ella proporcionan el enlace estructural entre el citoesqueleto y la matriz extracelular del miocito, conectando las proteínas contráctiles con el sarcolema, las cuales, al alterarse, producen fragilidad de la membrana que lleva a necrosis miofibrilar y pérdida de las fibras musculares con posterior reemplazo fibroadiposo. Otras distrofias musculares, como las de cintura-miembro tipo 2C-2F y tipo 2I, afectan también el complejo distrofina-glucoproteínas2. Estas patologías con alteraciones en este complejo se asocian a miocardiopatía dilatada como manifestación cardiaca principal1.
En comparación con las anteriores, las distrofias miotónica tipo1 y 2, la distrofia muscular de Emery Dreifuss, la distrofia muscular de cintura-miembro tipo1B y la distrofia fascioescapulohumeral se asocian con degeneración del sistema de conducción y arritmias como manifestación principal, además de compromiso miocárdico difuso variable1.
La distrofia miotónica es la enfermedad hereditaria del sistema neuromuscular más común en los pacientes adultos. Se trata de un trastorno multisistémico autosómico dominante con compromiso neurológico, muscular esquelético, cardiaco y liso, cuyas manifestaciones clínicas más llamativas son la debilidad distal y la miotonía. Adicionalmente se asocia a numerosas manifestaciones sistémicas, dentro de las cuales las más frecuentes son las cataratas, la diabetes mellitus, la hipogammaglobulinemia, el retardo en desarrollo psicomotor y la afección cardiaca3. Desde el punto de vista genético se caracteriza por una ampliación inestable del trinucleótido citosina-timina-guanina en la región no codificante 3′ del gen DMPK (dystrophia myotonica protein kinase), localizado en el brazo largo del cromosoma19. Tiene una prevalencia de 1:8.000 nacimientos y se presenta de 2 formas: el tipo1 (DM1 o distrofia de Steinert) o el tipo2 (DM2). La DM1 es la más común, y desde el punto de vista clínico se divide en 3 variedades según la edad de inicio: la congénita, la mínima y la clásica. La forma clásica es la más común, los síntomas se hacen evidentes entre la segunda y cuarta décadas de la vida, con progresión lenta en el tiempo, mientras que la forma congénita conlleva el peor pronóstico, con muerte en el período neonatal, la variedad mínima es de comienzo tardío luego de los 50años de edad3. Para la distinción entre la DM1 y la DM2 se han descrito cambios patológicos que podrían ser de utilidad, aunque el estudio genético es la prueba de oro. Se propone que la atrofia marcada de las fibras tipo1 es un cambio altamente indicativo de la DM14. En el presente caso, tanto las fibras tipo1 como las fibras tipo2 mostraron atrofia e hipertrofia y en forma similar internalización de los núcleos, sin que se notasen diferencias de proporción entre ambos tipos de fibras.
El compromiso cardiaco es una de las principales manifestaciones sistémicas en la DM1. El 30% de las muertes de estos pacientes se debe a complicaciones cardiacas y representa la segunda causa de mortalidad después de los problemas respiratorios. La biopsia endomiocárdica muestra cambios inespecíficos, como la fibrosis intersticial perivascular, la infiltración de grasa, la hipertrofia de miocardiocitos y la miocarditis focal. Existe una relación entre el número de repeticiones del triplete CTG, la edad de aparición de complicaciones cardiacas y el riesgo de episodios cardiacos mayores3.
Las manifestaciones cardiacas vistas en los pacientes con DM1 se pueden agrupar en 3 categorías: los trastornos de conducción, las taquiarritmias y otras anormalidades.
Trastornos de la conducciónLos trastornos de conducción ocurren hasta en el 40% de los pacientes y son la principal manifestación cardiaca. El sistema de conducción se puede afectar a cualquier nivel, pero es más común en el His-Purkinje. Se pueden encontrar trastornos leves en el electrocardiograma o en el estudio electrofisiológico (EEF), tales como prolongación del PR, QRS e intervalo His-ventricular (HV) (50% de casos) aun en pacientes asintomáticos. Estos trastornos pueden progresar a defectos graves y condicionar síntomas como la disnea, el mareo, el síncope y la muerte súbita. Debido a la naturaleza progresiva del compromiso cardiaco se recomienda seguimiento electrocardiográfico cada 6 a 12meses5. No existe claridad sobre la velocidad de progresión de los trastornos de conducción, mientras algunos autores reportan progresión lenta, otros relacionan la velocidad de progresión con el número de repeticiones de los trinucleótidos.
Los potenciales tardíos pueden ser positivos y manifiestan el retardo en la activación miocárdica causada por tejido anormal (fibrótico o necrótico). No obstante, este retardo en la activación miocárdica se observa a través de todo el sistema His-Purkinje de una manera homogénea y no solo a través de las áreas de la fibrosis, por ello son expresión del trastorno de conducción y representan un indicio de prolongación del HV. La duración del QRS>100ms y la presencia de potenciales tardíos predicen un HV prolongado con buena sensibilidad (80%) y especificidad (83%)6. Nuestro paciente no fue llevado a EEF ya que tenía flutter auricular con respuesta ventricular no controlada y QRS estrecho.
TaquiarritmiasLas taquicardias supraventriculares (sostenidas o no) son frecuentes hasta en el 25% de los casos, y la mayoría son asintomáticas. Las más frecuentes son flutter auricular (FlA) o fibrilación auricular (FA)2. En una cohorte de 161 pacientes, seguidos en promedio 5,1años, se identificó en 17FlA o FA7. Los factores de riesgo fueron la edad >46años, el género masculino, el electrocardiograma anormal y la FEVI disminuida, adicionalmente se encontró que la presencia de FA o de FlA se asoció, junto con la edad, a mayor mortalidad. En nuestro paciente su arritmia principal fue FlA.
También, pueden presentar arritmias ventriculares como la taquicardia ventricular (TV) o la fibrilación ventricular (FV). Los mecanismos por los cuales se puede generar una TV son la reentrada rama-rama y la reentrada alrededor de áreas de degeneración fibroadiposa del miocardio. La identificación de una TV por reentrada rama-rama es importante debido a que se puede ofrecer ablación por radiofrecuencia como tratamiento8. En pacientes con clínica de TV como el presíncope, el síncope, la disnea o la historia familiar de muerte súbita se aconseja realizar el EEF por el riesgo de arritmias fatales en edades tempranas.
Otras manifestacionesAunque los trastornos de la conducción y las arritmias son las manifestaciones cardiacas más frecuentes, se han descrito otras manifestaciones en pacientes con DM1. Un estudio mostró que el 1,8% de los pacientes con DM1 tienen signos y síntomas de falla cardiaca, dato quizás subestimado dada la inactividad física o la imposibilidad de comunicarlo por retardo en desarrollo psicomotor. Otras series han encontrado que menos del 10% de los pacientes tienen signos clínicos de falla cardiaca3. Sin embrago, el ecocardiograma o la resonancia magnética revelan alteraciones en porcentajes que oscilan desde el 33 al 78%. Se ha descrito hipertrofia ventricular izquierda y derecha, dilatación ventricular derecha, disminución de la FEVI del ventrículo izquierdo, infiltración de grasa, fibrosis en el ventrículo derecho o biventricular y prolapso de la válvula mitral.
La miocardiopatía dilatada no es común en la DM1, lo cual sugiere que el mecanismo de toxicidad generado por el ARN puede ser variable de órgano a órgano entre el músculo estriado esquelético, el cardiaco y el sistema de conducción. Otras manifestaciones cardiacas incluyen cardiopatía isquémica y prolapso de la válvula mitral por disfunción papilar (25 a 40%)8.
Los pacientes con DM1 tienen alto riesgo de muerte súbita, ya sea por bloqueos AV de alto grado o taquiarritmias ventriculares, en el 3,22% de los casos se requiere implante de marcapasos o cardiodesfibrilador para prevenir dichos episodios9. Un estudio encontró que los pacientes con DM1 que presentan alteraciones electrocardiográficas como intervalo PR igual o mayor a 240ms, QRS>120ms o taquiarritmias auriculares tienen mayor riesgo de muerte súbita. Se encontró, además, que la muerte súbita es más frecuente en los pacientes con edad avanzada y con mayor compromiso muscular, lo cual sugiere estadios más avanzados de la enfermedad9.
Nuestro paciente tuvo una presentación cardiaca inicial no típica de DM1, con falla cardiaca con FlA pero sin trastorno de conducción. No obstante, las manifestaciones musculares eran altamente compatibles con el diagnóstico y nos deben alertar hacia la búsqueda de compromiso muscular periférico en pacientes con compromiso miocárdico sin causa aparente, pacientes jóvenes o con antecedentes familiares de cardiopatía o muerte súbita temprana. En este caso, el diagnóstico de DM1 fue basado en la impresión clínica, la electromiografía y la biopsia. Es importante resaltar las limitaciones para realizar estudios genéticos moleculares en nuestro medio, lo que dificulta la posibilidad de consejería no solo del individuo afectado, sino también de su familia.
ConclusionesSe trata de un caso clínico de DM1, una patología infrecuente, teniendo en cuenta que muchas de las enfermedades neuromusculares hereditarias afectan el corazón y son responsables de una proporción significativa de la morbimortalidad en esta patología.
Una atención adecuada de esta patología requiere la formación de equipos multidisciplinarios con un abordaje dinámico y de manera conjunta de cardiólogos y neurólogos, adaptándose a los requerimientos particulares de cada caso individual.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.