


El balance entre agentes vasoconstrictores y vasodilatadores, así como factores mitogénicos y antimitogénicos derivados del endotelio, está alterado en algunas situaciones y trae como resultado final un aumento en la presión arterial pulmonar. La disfunción endotelial es promovida por estímulos como hipoxia, acidosis, radicales libres, mediadores inflamatorios, tensión tangencial causada por aumento del flujo sanguíneo pulmonar de izquierda a derecha por cortocircuito intracardiaco y fibrina derivada de tromboembolia. La disfunción endotelial y el remodelado vascular son dos procesos importantes que explican el desarrollo de hipertensión pulmonar. El enfoque terapéutico de esta entidad ha progresado rápidamente en los últimos años, pero aún no existe un tratamiento ideal. Estrategias para el futuro pueden incluir mejoría en los métodos para administrar los medicamentos disponibles, combinaciones de los mismos, nuevos grupos terapéuticos y la posibilidad de terapia genética.
The equilibrium between vasoconstrictors and vasodilators, as well as mitogenic and anti-mitogenic factors originating from the endothelium, is upset in certain situations and as a final result leads to an increase in pulmonary arterial pressure. The endothelial dysfunction is promoted by stimulants such as hypoxia, acidosis, free radicals, inflammatory mediators, tangential pressure caused by an increase in pulmonary blood flow from the left to the right by an intra-cardiac short circuit and fibrin derived from embolisms. Endothelial dysfunction and vascular remodelling are two important processes that explain the development of pulmonary hypertension. The therapeutic approach to this condition has progressed rapidly in the last few years, but there is still no ideal treatment. Future strategies could include improvements in the methods for administering the medications available, combinations of these, new therapeutic groups, and the possibility of gene therapy.
La hipertensión arterial pulmonar es una enfermedad devastadora, que es precipitada por el remodelado hipertrófico vascular pulmonar de las arteriolas distales, para así aumentar la presión arterial pulmonar y la resistencia vascular. Esto debe suceder en ausencia de falla cardiaca izquierda, enfermedad parenquimatosa pulmonar o enfermedad tromboembólica. A pesar de la terapia médica disponible, el remodelado de la arteria pulmonar y sus consecuencias hemodinámicas se traducen en disfunción ventricular derecha, falla cardiaca y muerte temprana. Para limitar la morbilidad y mortalidad, se ha enfocado la atención en descubrir los mecanismos celulares y moleculares subyacentes al remodelado aberrante de la arteria pulmonar para identificar sus vías y así mediar una intervención. Existe un reconocido componente genético heredable para desarrollar hipertensión arterial pulmonar, aunque también hay evidencia de otras alteraciones genéticas, incluyendo un daño en el ADN celular vascular pulmonar, activación de una respuesta dañina del ADN y variaciones en expresión del microARN. Estos hallazgos probablemente contribuyen a la regulación inadecuada de la proliferación y apoptosis siguiendo vías similares a las que se observan en cáncer. También aparecen cambios en el metabolismo celular, flujo metabólico y función mitocondrial, así como la transición mesenquimal a endotelial como clave para las vías y señales que promueven el remodelado vascular pulmonar1.
Las células endoteliales son reconocidas como los mayores reguladores de la función pulmonar, de modo que la disfunción endotelial llega a ser la causa del disbalance en la producción endotelial de vasoconstrictores vs. vasodilatadores, así como activadores vs. inhibidores de la célula muscular lisa, su crecimiento y migración. También regulan la homeostasis entre mediadores protrombóticos y antitrombóticos, así como señales proinflamatorias vs. antiinflamatorias2.
La circulación pulmonar es un sistema de baja presión y alto flujo con una gran capacidad para reclutamiento de los vasos normalmente no perfundidos. Como consecuencia, las paredes de las arterias pulmonares son delgadas por su baja presión transmural. La hipertensión arterial pulmonar es una enfermedad de las arterias pulmonares pequeñas, caracterizada por un estrechamiento progresivo que lleva a un aumento en la resistencia vascular pulmonar. La consecuencia es un aumento de la poscarga del ventrículo derecho y su final falla. La vasocontricción, el remodelado vascular y la trombosis contribuyen al aumento en la resistencia vascular pulmonar3. Como sea, ahora se reconoce que la obstrucción arterial pulmonar por proliferación y remodelado es clave en su patogénesis4. Los mecanismos fisiopatológicos descritos están presentes en mayor o menor grado en todos los tipos de hipertensión pulmonar pero compartiendo mayores similitudes cuando pertenecen al mismo grupo de clasificación de hipertensión pulmonar. Los cinco grupos clínicos de clasificación de la hipertensión pulmonar comparten hallazgos similares patológicos, hemodinámicos y por ende un enfoque terapéutico similar5.
Fisiopatología de la hipertensión arterial pulmonar en algunas entidades específicasAlgunas patologías específicas comparten características comunes, máxime si pertenecen al mismo grupo clínico de clasificación, pero de la misma forma pueden existir diferencias marcadas y muchos de estos aspectos no están del todo esclarecidos. En la hipertensión arterial pulmonar asociada a virus de inmunodeficiencia humana, el mecanismo se entiende por completo y no se ha documentado la acción directa del virus en el endotelio. Es claro el papel de la predisposición genética, las proteínas totales que inducen estrés oxidativo, la proliferación del músculo liso, el daño endotelial, la inflamación mediada por citoquinas, la vasoconstricción, el remodelado vascular y los eventos microtrombóticos6,7. En esclerosis sistémica, entidad en la cual la hipertensión arterial pulmonar es una complicación vascular devastadora, la inflamación y la autoinmunidad desempeñan un rol significativo en su desarrollo, por niveles elevados de interleuquinas 1 y 6, así como P-selectina8. El papel de la autoinmunidad se sugiere por la presencia de un número de autoanticuerpos en suero de pacientes con esclerosis sistémica como anticentrómero, antitopoisomerasa I, anti-ARN-polimerasa III, anti-fibrilarina y anti-TH/TO9. En hipertensión portal, la fisiopatología no está bien definida y el desarrollo de la hipertensión pulmonar es independiente de la causa de la hipertensión portal y de la gravedad de la misma10,11. Los factores principales involucrados son la vasoproliferación, la resistencia aumentada al flujo arterial y la disminución en la expresión de las prostaciclinas en las arterias pulmonares12. El factor genético que participa en la hipertensión arterial pulmonar idiopática no se ha demostrado en hipertensión portal. Es importante señalar que sujetos genéticamente susceptibles pueden desencadenar la enfermedad como respuesta a señales intracelulares. A nivel arteriolar, es posible apreciar arteriopatía plexiforme, hipertrofia de la media, fibrosis de la íntima, proliferación de la adventicia y necrosis fibrinoide. Asociado a lo anterior, se han documentado trombos y recanalización que se traducen en trombosis in situ, causada por daño endotelial, agregación plaquetaria y cierto estado de hipercoagulabilidad concomitante13. En hipertensión pulmonar tromboembólica crónica, la extensión de la obstrucción vascular es el mayor determinante de la severidad de esa hipertensión. En la mayoría de casos, más del 40% del lecho vascular pulmonar está obstruido; el empeoramiento de la hipertensión pulmonar puede deberse a tromboembolia recurrente o trombosis in situ y remodelado de las arterias pulmonares distales en áreas no ocluidas, similar a lo que ocurre en hipertensión pulmonar idiopática. Varias líneas de evidencia apoyan esta hipótesis:
- 1.
Baja correlación entre la extensión de la obstrucción central y el grado de la hipertensión pulmonar.
- 2.
Progresión documentada de la hipertensión pulmonar sin tromboembolia recurrente.
- 3.
Evidencia de redistribución del flujo vascular pulmonar después de tromboendartectomía de las áreas no ocluidas a las áreas sometidas a endarterectomía, como resultado de la resistencia vascular pulmonar aumentada en el lecho vascular no obstruido.
- 4.
Evidencia histopatológica de vasculopatía pulmonar con hipertrofia de la media, engrosamiento de la íntima y lesiones plexiformes.
- 5.
Hipertensión pulmonar persistente a pesar de una tromboendartectomía satisfactoria en el 10% de los pacientes14–16.
En conclusión, la falta de correlación entre la proporción de las arterias pulmonares obliteradas y las cifras de hipertensión pulmonar, sugiere que una teoría exclusivamente mecánica, podría ser demasiado simplista. En embolia pulmonar aguda podría ser el evento inicial, pero la progresión de la enfermedad resultaría del remodelado vascular progresivo de los pequeños vasos. Es posible que la trombosis arterial pulmonar no resuelta sea un factor decisivo para que células endoteliales vasculares inicien su transición mesenquimal. Es decir, aspectos genéticos, mecanismos inflamatorios y condiciones clínicas influyen en el mosaico fisiopatológico de esta compleja entidad17.
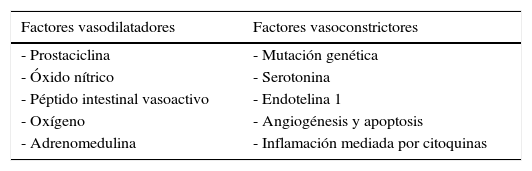
Habiendo señalado algunos aspectos importantes de condiciones clínicas específicas, se revisarán en forma general algunos de los factores que afectan los fenómenos de autorregulación conducentes al mantenimiento de la constancia, propiedades u homeostasis de la hemodinamia pulmonar. Es decir, el balance entre factores vasoconstrictores y vasodilatadores comunes a todas las entidades relacionadas con hipertensión arterial pulmonar18,19. En la tabla 1 se resumen los elementos desencadenantes más importantes implicados en la pérdida de la homeostasis vascular.
Factores implicados en la homeostasis de la hemodinamia pulmonar
| Factores vasodilatadores | Factores vasoconstrictores |
|---|---|
| - Prostaciclina | - Mutación genética |
| - Óxido nítrico | - Serotonina |
| - Péptido intestinal vasoactivo | - Endotelina 1 |
| - Oxígeno | - Angiogénesis y apoptosis |
| - Adrenomedulina | - Inflamación mediada por citoquinas |
Se han identificado más de 300 mutaciones independientes en el BMPR1 (receptor de la proteína morfogénica ósea), como causa de aproximadamente 75% de los casos de hipertensión arterial pulmonar familiar y por encima del 25% de los casos aparentemente esporádicos, de modo que se ha establecido que el defecto en este gen es el mayor determinante genético para hipertensión arterial pulmonar. Se ha determinado, igualmente, una mutación patológica en el receptor I ACBRL1 y en mayor frecuencia el receptor tipo III endoglina que se asocia con telangiectasia hemorrágica hereditaria. Estas observaciones avalan un papel predominante del factor transformante de crecimiento beta como una súper familia de receptores involucrados en el crecimiento, reconocimiento y diferenciación celular, así como en la apoptosis. El BMPR1 mediante la interacción con segundos mensajeros, que son las proteínas SMAD, realiza las transducción de señales para el remodelado y la apoptosis endotelial. El gen BMPR2 situado en el brazo largo del cromosoma 2 (dosq33-q34), codifica una proteína denominada receptor tipo II de la proteína morfogénica ósea. Este gen ejerce un papel en la regulación del número de células en ciertos tejidos. La proteína codificada por este gen se sitúa a través de la membrana celular, de manera que un extremo de la proteína está en la superficie externa de la célula y el otro permanece en el interior. Esta disposición hace que la proteína reciba y transmita señales que ayudan a la proliferación celular o la apoptosis. Este proceso regula el número de células en los tejidos. Se han identificado más de 140 mutaciones en el gen BMPR2 en hipertensión arterial pulmonar. Alrededor de la mitad de estas mutaciones interrumpen el montaje del receptor tipo II de la proteína morfogénica ósea, reduciendo la cantidad de esta en las células. Otras mutaciones impiden que la proteína alcance la superficie celular o alteren su estructura, de modo que no pueden recibir ni transferir señales. Se ha identificado también una mutación genética en la caveolina 1 (CAV1), la cual codifica una proteína de membrana abundante en el endotelio y otras células del pulmón. Estas proteínas son abundantes en la superficie celular y sus receptores son críticos para iniciar señales de cascada como la súper familia del factor de crecimiento transformante beta, la vía del óxido nítrico y la proteína G. De otro lado, señales aberrantes en la membrana plasmática pueden ser el mecanismo de patogénesis de hipertensión arterial pulmonar20,21.
ProstaciclinaEs un vasodilatador pulmonar endógeno que actúa a través de la activación del adenosín monofosfato cíclico (AMPC). También inhibe la proliferación de células vasculares musculares lisas y disminuye la agregación plaquetaria. La síntesis de prostaciclina está disminuida en las células endoteliales de pacientes con hipertensión arterial pulmonar. De esta forma, la terapia con prostaciclina ha demostrado mejoría en la hemodinamia, condición clínica y sobrevida de pacientes con hipertensión arterial pulmonar22,23.
Óxido nítricoLa alteración en el endotelio derivada en falta de vasodilatación se ha avalado por la demostración de una reducción de la óxido nítrico sintetasa, expresada en la células del endotelio vascular pulmonar. Una estrategia terapéutica en hipertensión arterial pulmonar consiste en el aumento de óxido nítrico, mediado por guanosín monofosfato cíclico (GMPC) por la inhibición de su rompimiento por la fosfodiesterasa tipo 524.
Péptido intestinal vasoactivoEs un neuropéptido que funciona primariamente como neurotransmisor y actúa como vasodilatador pulmonar sistémico potente. También inhibe la proliferación de células vasculares musculares lisas y disminuye la agregación plaquetaria. Interviene a través de dos subtipos de receptores (VPAC-1 y 2), que se acoplan con adenilciclasa y se expresan en la vasculatura pulmonar. La estimulación de estos receptores lleva a la activación de GMPC. Bajas concentraciones séricas de péptido intestinal vasoactivo se observan en las arterias pulmonares de pacientes con hipertensión arterial pulmonar25.
SerotoninaEn hipertensión arterial pulmonar los niveles de serotonina circulantes están elevados, mientras que el mayor depósito de serotonina, la 5-hidroxitriptamina (5-HT) está baja. La 5-HT se produce en las células enterocromafines del tracto gastrointestinal así como en los cuerpos neuroepiteliales pulmonares y se almacena en las plaquetas. Se ha propuesto un papel importante de la 5-HT en la hipertensión arterial pulmonar. Una correlación entre altos niveles en plasma de 5-HT, se ha observado en pacientes con hipertensión arterial pulmonar. Esto no se ha corregido con el tratamiento con epoprostenol ni trasplante, indicando que el aumento de 5-HT no es solo una consecuencia de la presión de la arteria pulmonar. La expresión de la producción de la 5-HT en las células musculares arteriales lisas contribuye al remodelado vascular pulmonar que ocurre en hipertensión arterial pulmonar clínica y experimental. En la década de 1960 se encontró una asociación entre hipertensión arterial pulmonar y el anorexígeno aminorex. Este último indujo liberación de 5-HT e inhibición de monoamino oxidasa, disminuyendo potencialmente su metabolismo para así aumentar los niveles plasmáticos de 5-HT26,27.
Endotelina 1Existe fuerte evidencia que afirma que la endotelina 1 (ET-1) derivada del endotelio, tiene un papel principal en el disbalance vasoconstrictor/vasodilatador característico de la hipertensión arterial pulmonar. Los niveles pulmonares y circulantes de ET-1 están aumentados en animales y pacientes con hipertensión arterial pulmonar de cualquier etiología. Estas observaciones demuestran que la ET-1 contribuye al componente vasoactivo, así como el remodelado vascular pulmonar característico de esta condición. Esto ubica la terapia antagonista del receptor de la endotelina 1 como de gran relevancia en hipertensión arterial pulmonar28.
Angiogénesis y apoptosisEl factor de crecimiento endotelial vascular (VEGF) y dos receptores tirosín quinasa de gran afinidad, angiogénicos específicos celulares endoteliales (VEGFR-1 y VEGFR-2), desempeñan un rol en hipertensión arterial pulmonar. Aunque se desconce el papel fisiológico de la expresión aumentada de VEGF en el pulmón, se ha propuesto que avala la sobrevida y el mantenimiento de la célula endotelial. En hipertensión arterial pulmonar la expresión de VEGF está aumentada dentro de la vasculatura pulmonar, incluyendo las lesiones plexiformes. Así mismo, en hipertensión arterial pulmonar idiopática la expresión VEGF-1 está elevada. En ratas se ha observado que la combinación de un bloqueo crónico VEGF-2 e hipoxia crónica puede causar disfunción celular endotelial pulmonar y muerte celular, permitiendo la aparición de una resistencia a la proliferación y apoptosis celular y desarrollo posterior de hipertensión pulmonar severa29,30.
AdrenomedulinaEs un potente vasodilatador que se secreta en varios tejidos incluidos corazón y pulmón. Con su administración se ha observado vasodilatación, así como un efecto sobre la angiogénesis y la apoptosis31.
ConclusionesPara el desarrollo de hipertensión arterial pulmonar debe existir una predisposición genética debida a algunas mutaciones, o puede haber algún tipo de interacción ambiental como anorexígenos, agentes tóxicos, infección por el tipo de inmunodeficiencia humana, enfermedad del colágeno o hipertensión portal, entre otras. Por otro lado, pueden presentarse otro tipo de noxas o de expresión de genes como el aumento de la serotonina, alteración en el transporte de la serotonina, disminución del óxido nítrico y de la prostaciclina, y aumento de la endotelina o del tromboxano que a su vez se traducen en un incremento en la producción de elastasa y matriz de proteinasa, que produce elevación del colágeno y de la elastina. Recientemente se han encontrado cambios inflamatorios en las células del endotelio de las arterias pulmonares, que podrían contribuir a la patogenia.
Conflicto de interesesNinguno.