




Poco se ha estudiado acerca del comportamiento de la hipertensión pulmonar en pediatría en población peruana, incluso en el mundo, la información es deficiente, de ahí la necesidad de desarrollar estudios y registros para la orientación del diagnóstico y tratamiento oportuno.
Material y métodosEstudio retrospectivo, observacional, transversal, en el que se incluyeron pacientes de 3 meses hasta 15 años de edad, con diagnóstico de hipertensión pulmonar, sometidos a estudio hemodinámico por cateterismo cardíaco en INCOR, entre enero de 2015 a diciembre de 2016 (2 años).
ResultadosSe diagnosticaron 57 pacientes con hipertensión pulmonar, 51% varones, de los cuales 22% procedían de una altura mayor 2.500 metros sobre el nivel del mar. Grupo etario predominante 1-3 años. Clase funcional II 49%. Síndrome de Down (16%). La comunicación interventricular / ducto arterioso persistente (CIV / PCA) fue la cardiopatía más frecuente. El grupo 1(NICE2013) fue el más frecuente con 51 casos (89,5%); dentro de éste resalta el grupo asociado a cardiopatías congénitas (48 casos), mientras que en el grupo 2 se clasificaron 6 casos (10,5%). La presión arterial pulmonar media más frecuente fue mayor 40 mm Hg, con aumento de RVPi y RVP/ RVS, leve a moderado. Se realizaron pruebas de vasorreactividad en 14 pacientes (24%), la cual fue positiva en 12 de 14 casos. En el 52% de los casos se decidió la reparación quirúrgica del defecto.
ConclusiónEste estudio constituye el primer registro de hipertensión pulmonar en niños peruanos en el que se halló que aquellos con esta enfermedad presentan características propias, según los distintos grupos; así mismo, la asociación a cardiopatías congénitas fue importante. Se recomienda el tratamiento oportuno y pronóstico, ya que el compromiso vascular puede estar presente en estadios clínicamente no significativos.
There have been few studies on the behaviour of pulmonary hypertension in paediatrics in the Peruvian population. Even in the world, there is insufficient information. Thus, there is a need to develop studies and registers in order to focus on a timely diagnosis and treatment.
Material and methodsA retrospective, observational and cross-sectional study was performed. Patients from 3 months to 15 years of age and with a diagnosis of pulmonary hypertension, and subjected to a haemodynamic study by cardiac catheterisation in INCOR (National Cardiovascular Institution) were included between January 2015 and December 206 (2 years).
ResultsOut of a total of 57 patients diagnosed with pulmonary hypertension, 51% were males of whom 22% came from an altitude greater than 2,500 metres above sea level. The age group were predominantly from 1 -3 years. Just under half (49% were functional class II, and 16% had Down's syndrome. Left ventricular growth (LVG) +/- Patent ductus arteriosus (PDA) was the most frequent cardiac disease. Group 1 1(NICE 2013) was the most frequent with 51 (89.5%) cases. Within this, the group associated to congenital heart disease (48 cases) is highlighted, while 6 cases (10.5%) were classified in Group 2. The most frequent mean pulmonary arterial pressure was greater than 40 mm Hg, with a mild to moderate increase in indexed pulmonary vascular resistance (iPVR and pulmonary vascular resistance (PVR)/ pulmonary vascular resistance (PVS). Vasoreactivity tests were performed on 14 (24%) patients, which was positive in 12 of the 14 cases. Surgical repair of the defect was decided in 52% of cases.
ConclusionThis study forms the first register of pulmonary hypertension in Peruvian children, in which it was found that children with this disease have their own characteristics, according to the different groups. Likewise, the relationship with congenital cardiac disease was important. Timely and prognostic treatment is recommended, since the vascular compromise can be present clinically non-significant states.
La hipertensión pulmonar es una causa importante de morbilidad y mortalidad en la población pediátrica. En su etiología se incluyen las cardiopatías congénitas (cuya prevalencia es 6 a 8 por 1.000 recién nacidos vivos1,2. Las cardiopatías congénitas tienen presentación heterogénea, y su evolución y ritmo de progresión dependerán de factores como tipo, tamaño y tiempo en el que se decide corregir el defecto, además de mutaciones genéticas definidas, enfermedad pulmonar de diversa etiología y enfermedades hematológicas concomitantes3.
Es indispensable destacar algunos aspectos fisiopatológicos de la hipertensión pulmonar en pediatría que la diferenciarán de aquella de la edad adulta. La presión pulmonar es similar a la presión arterial sistémica in útero, pero rápidamente cae después del nacimiento, llegando a los dos a tres meses de edad postnatal a ser similar a la de los adultos4–6.
La clasificación clínica de la hipertensión pulmonar ha pasado por una serie de cambios desde la primera clasificación propuesta en 1973 en la Conferencia Internacional sobre hipertensión pulmonar primaria en Ginebra (Suiza), hasta las actuales7. En relación con la hipertensión arterial pulmonar asociada a cardiopatías congénitas, las guías europeas la dividen en cuatro grupos6–8.
En el momento del diagnóstico inicial de hipertensión pulmonar, la historia clínica y el examen físico, complementados con pruebas diagnósticas no invasivas como el ecocardiograma son importantes para la detección de hipertensión pulmonar, en tanto que para confirmar el diagnóstico y evaluar la gravedad de la alteración hemodinámica se requiere del cateterismo del corazón derecho, el cual debe hacerse en un centro experimentado antes de la iniciación de terapia dirigida a la hipertensión arterial pulmonar. Las excepciones pueden incluir enfermedades graves o pacientes que requieren el inicio inmediato de la terapia9.
El cateterismo cardíaco debe incluir pruebas de vasorreactividad aguda. Se han utilizado diversos tratamientos farmacológicos durante su realización. El objetivo es definir la presencia de vasorreactividad pulmonar, destacando el oxígeno al 100%, el isoproterenol, la adenosina, y en épocas recientes, el óxido nítrico y el iloprost, a menos que exista una contraindicación. El oxígeno al 100% se usa específicamente en la altura, mientras que en niños el más utilizado para el test de vasorreactividad es el óxido nítrico.
Estas pruebas ayudan para definir la operabilidad, sin embargo también deben considerarse la edad del paciente, el tipo de lesión y los hallazgos clínicos10–12.
En Perú el diagnóstico de hipertensión arterial pulmonar se realiza principalmente en centros especializados y la mayoría de los estudios se llevan a cabo en el Instituto Nacional del Corazón (INCOR), que es el único centro de referencia especializado del país, para los pacientes con seguro social, donde se realizan en promedio 320 cateterismos cardiacos por año y de éstos el 51% está conformado por los cateterismos terapéuticos (intervencionistas) y el 49% los cateterismos diagnósticos (estudios hemodinámicos); los resultados de estos últimos fueron la fuente de datos de este estudio. En nuestro medio es desconocida la caracterización clínica y epidemiológica de la hipertensión arterial pulmonar y no se han hecho trabajos de investigación en este grupo etario. Por tanto, el objetivo fue conocer el comportamiento clínico, la etiología y los hallazgos de los parámetros hemodinámicos de esta enfermedad en nuestra población pediátrica, constituyendo así el primer registro en niños peruanos.
Material y métodosEstudio retrospectivo, observacional, transversal, en el que se incluyeron todos los pacientes mayores de tres meses de vida y menores de 15 años, con diagnóstico de hipertensión pulmonar que fueron sometidos a estudio hemodinámico por cateterismo cardíaco en INCOR, durante enero de 2015 a diciembre de 2016. Para el análisis de los resultados se empleó estadística descriptiva, programa de Excel 2010. Se determinó hipertensión pulmonar si se cumplían los siguientes criterios: pacientes mayores de 3 meses de vida, presión media de la arteria pulmonar superior a 25mm Hg a nivel del mar; se hace referencia a hipertensión arterial pulmonar cuando se agrega la presión en cuña de la arteria pulmonar (PCAP) o la presión diastólica final del ventrículo izquierdo(PDFVI) < o igual a 15mm Hg, y en cuanto al parámetro hemodinámico si la resistencia vascular pulmonar indexada (RVPi) es mayor a 3 UW/m22,3.
Todos los cateterismos se hicieron bajo anestesia general mixta inhalatoria (con tubo endotraqueal o mascarilla laríngea) y endovenosa, con FiO2 menor o igual al 30%. En el estudio hemodinámico se colocaron introductores venosos y arteriales (se administró heparina a la dosis de 50 a 100 unidades por kilogramo de peso). Se tomaron muestras para oximetrías y se realizó registro de presiones para los cálculos hemodinámicos y la valoración de hipertensión arterial pulmonar.
Las pruebas de reactividad pulmonar (PVA) se llevaron a cabo cuando las presiones arteriales pulmonares medias tenían valores altos, compatibles con hipertensión pulmonar severa, para evaluar la respuesta vascular pulmonar a vasodilatadores pulmonares. Se realizaron pruebas farmacológicas (dependiendo de la disponibilidad de la institución) y no farmacológicas como el test de hiperoxia con oxígeno al 100%, durante 10 minutos, luego de lo cual se procedió a tomar nuevamente muestras para oximetrías y registro de presiones repitiendo los cálculos hemodinámicos y evaluando la variación registrada respecto a la basal. En los casos de conducto arterioso persistente se procedió a la oclusión del defecto para evaluar su comportamiento hemodinámico. Se consideró respuesta positiva si ocurría una disminución de más del 20% de la presión arterial pulmonar media, RVPi y RVP / RVS o si se obtenían valores finales de RVPi <6 UW y RVPi / RVSi <0,34.
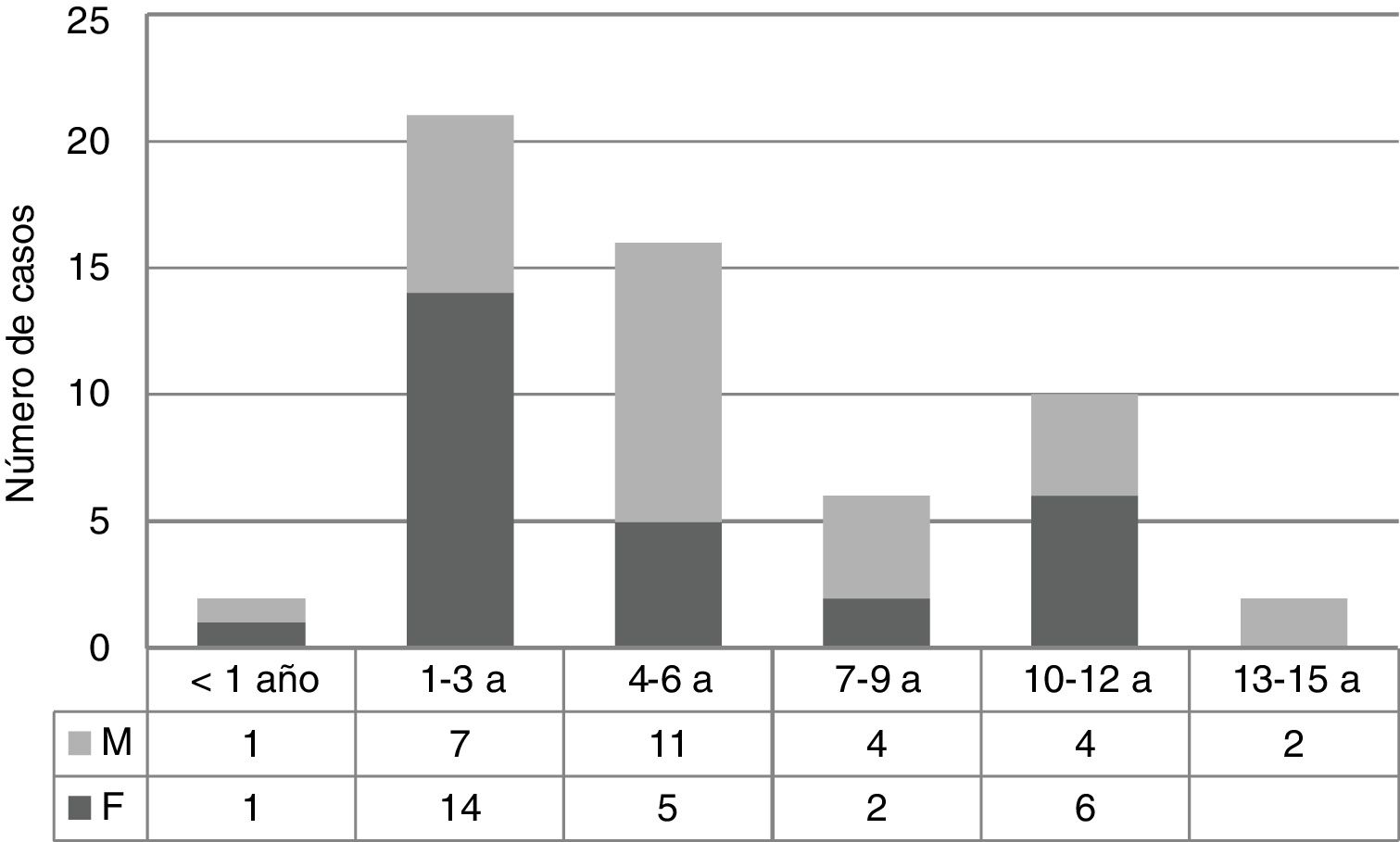
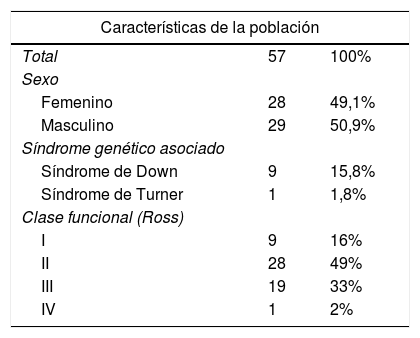
ResultadosEn la tabla 1 se identifican un total de 57 casos en los cuales no hubo diferencia en general entre ambos sexos. Se encontraron pacientes con trisomía 21 y síndrome de Turner y además la mayoría tenía algún grado de afectación de la clase funcional (según la escala de Ross). La edad más frecuente fue 3 años, con un rango de 3,5 meses de edad y 14 años, encontrándose hipertensión pulmonar más comúnmente en la edad entre 1 a 6 años (incluye los grupos 1-3 y 4-6 años) (fig. 1). La hipertensión pulmonar se clasificó según Nice 2013 (tabla 2); el 89,5% correspondió al grupo 1 (hipertensión arterial pulmonar) y el 10,5% al grupo 2. El subgrupo más frecuente del grupo 1 fue el asociado a cardiopatías congénitas (84,2%), el cual a su vez se subclasificó según la clasificación clínica2, donde predominó el grupo II con 42 casos (73,7%); el menos frecuente fue el grupo I con 2 casos (3,5%).
Características clínicas y demográficas del total de pacientes pediátricos con hipertensión pulmonar
| Características de la población | ||
|---|---|---|
| Total | 57 | 100% |
| Sexo | ||
| Femenino | 28 | 49,1% |
| Masculino | 29 | 50,9% |
| Síndrome genético asociado | ||
| Síndrome de Down | 9 | 15,8% |
| Síndrome de Turner | 1 | 1,8% |
| Clase funcional (Ross) | ||
| I | 9 | 16% |
| II | 28 | 49% |
| III | 19 | 33% |
| IV | 1 | 2% |

Clasificación de la hipertensión pulmonar
| Clasificación | N | F |
|---|---|---|
| Grupo 1: hipertensión arterial pulmonar | 51 | 89,5% |
| 1.1 Idiopática o 1.2 Hereditaria | 3 | 5,3% |
| 1.4 Hipertensión arterial pulmonar asociada a: | 0,0% | |
| 1.4.4 Cardiopatías congénitas (clasificación clínica): | ||
| I. Síndrome de Eisenmenger | 2 | 3,5% |
| II. Hipertensión arterial pulmonar asociada a cortocircuito sistémico-pulmonar | 42 | 73,7% |
| III. Hipertensión arterial pulmonar asociada con pequeños defectos septales | 0 | 0% |
| IV. Hipertensión arterial pulmonar postoperatoria | 4 | 7% |
| Grupo 2: hipertensión pulmonar debida a enfermedad cardíaca izquierda | 6 | 10,5% |
| 2.1 Disfunción sistólica del ventrículo izquierdo | 3 | 5,3% |
| 2.3 Enfermedad valvular | 2 | 3,5% |
| 2.4 Cardiopatías congénitas o adquiridas afectando la entrada o salida del ventrículo izquierdo y miocardiopatía congénita | 1 | 1,7% |
| Grupo 3: hipertensión pulmonar asociada a enfermedades pulmonares y/o a hipoxemia | 0 | 0,0% |
| Grupo 4: hipertensión pulmonar por enfermedad tromboembólica crónica | 0 | 0,0% |
| Grupo 5: hipertensión pulmonar con mecanismos multifactoriales no claros | 0 | 0,0% |
| Total de casos | 57 | 100,0% |
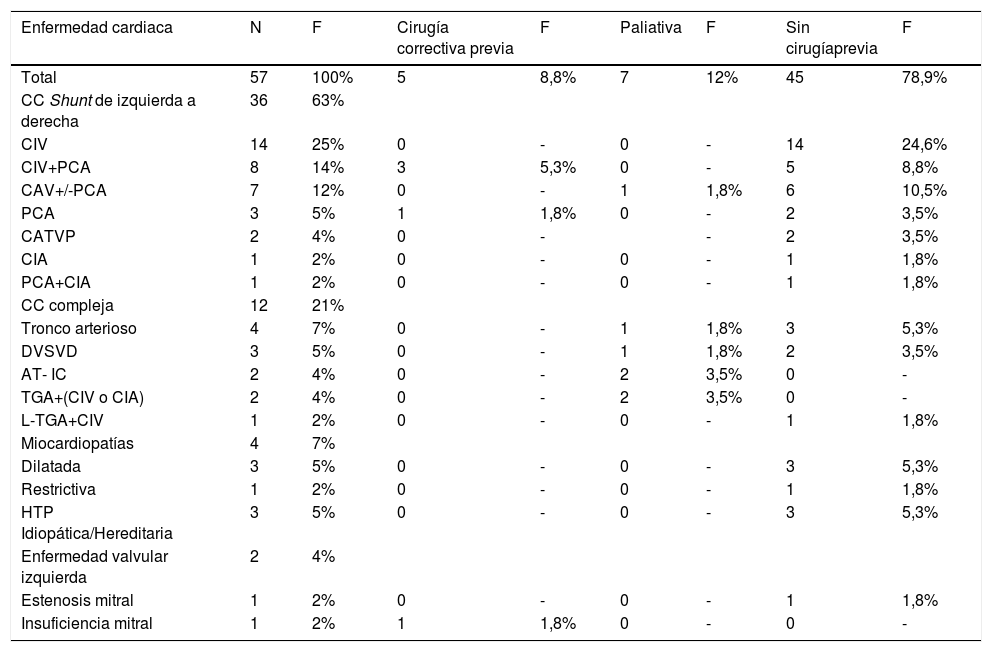
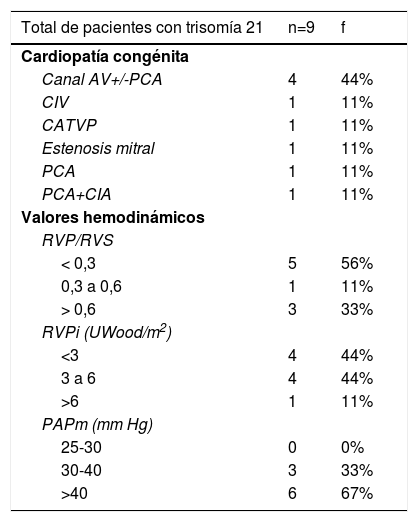
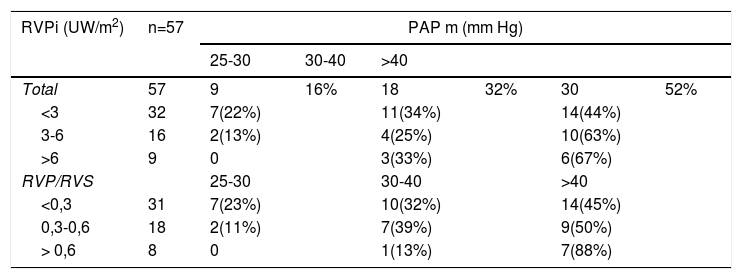
En cuanto a las distintas enfermedades cardíacas asociadas a hipertensión pulmonar (tabla 3) se halló predominio de las cardiopatías congénitas (88%), dentro de las cuales la comunicación interventricular fue la más frecuente (39%), bien fuera como defecto aislado o asociado a PCA, constituido por 22 pacientes de los cuales solo 3 habían sido operados. En la tabla 4 se muestran los resultados en los niños con trisomía 21, en cuyo caso la cardiopatía congénita más frecuente asociada a hipertensión arterial pulmonar fue el canal aurículo-ventricular con o sin PCA. En cuanto a hemodinamia, el 56% tenía valores altos de presión arterial pulmonar media y solo el 16% RVPi >6 UW/m2, considerado alto. En la tabla 5 se estudió la relación entre los valores hemodinámicos; se encontró que la mayor parte de los casos presentaba valores considerados altos PAP >40mm Hg (severo), y a su vez éstos en su mayor parte tenían resistencias pulmonares bajas: RVPi< 3 UW y RVP/RVS < 0,3. Cabe destacar que dentro del grupo con presión arterial pulmonar media 30-40mm Hg (moderado) hay casos con resistencias pulmonares altas: RVPi > 6 UW/m2 y RVP/ RVS> 0,6 (severo).
Enfermedad cardiaca asociada a hipertensión pulmonar y frecuencia de cirugías previas (correctiva o paliativa: banding pulmonar)
| Enfermedad cardiaca | N | F | Cirugía correctiva previa | F | Paliativa | F | Sin cirugíaprevia | F |
|---|---|---|---|---|---|---|---|---|
| Total | 57 | 100% | 5 | 8,8% | 7 | 12% | 45 | 78,9% |
| CC Shunt de izquierda a derecha | 36 | 63% | ||||||
| CIV | 14 | 25% | 0 | - | 0 | - | 14 | 24,6% |
| CIV+PCA | 8 | 14% | 3 | 5,3% | 0 | - | 5 | 8,8% |
| CAV+/-PCA | 7 | 12% | 0 | - | 1 | 1,8% | 6 | 10,5% |
| PCA | 3 | 5% | 1 | 1,8% | 0 | - | 2 | 3,5% |
| CATVP | 2 | 4% | 0 | - | - | 2 | 3,5% | |
| CIA | 1 | 2% | 0 | - | 0 | - | 1 | 1,8% |
| PCA+CIA | 1 | 2% | 0 | - | 0 | - | 1 | 1,8% |
| CC compleja | 12 | 21% | ||||||
| Tronco arterioso | 4 | 7% | 0 | - | 1 | 1,8% | 3 | 5,3% |
| DVSVD | 3 | 5% | 0 | - | 1 | 1,8% | 2 | 3,5% |
| AT- IC | 2 | 4% | 0 | - | 2 | 3,5% | 0 | - |
| TGA+(CIV o CIA) | 2 | 4% | 0 | - | 2 | 3,5% | 0 | - |
| L-TGA+CIV | 1 | 2% | 0 | - | 0 | - | 1 | 1,8% |
| Miocardiopatías | 4 | 7% | ||||||
| Dilatada | 3 | 5% | 0 | - | 0 | - | 3 | 5,3% |
| Restrictiva | 1 | 2% | 0 | - | 0 | - | 1 | 1,8% |
| HTP Idiopática/Hereditaria | 3 | 5% | 0 | - | 0 | - | 3 | 5,3% |
| Enfermedad valvular izquierda | 2 | 4% | ||||||
| Estenosis mitral | 1 | 2% | 0 | - | 0 | - | 1 | 1,8% |
| Insuficiencia mitral | 1 | 2% | 1 | 1,8% | 0 | - | 0 | - |
CAV: canal atrioventricular común, CIV: comunicación interventricular, CATVP: conexión anómala total de venas pulmonares, PCA: persistencia de conducto arterioso, CIA: comunicación interauricular, DSVD: doble vía de salida de ventrículo derecho, AT: atresia tricuspídea, TGA: transposición de grandes arterias, L-TGA: transposición corregida de grandes arterias.
Tipo de cardiopatías congénitas y clasificación según valores hemodinámicos: RVPi, RVP/RVS en pacientes con trisomía 21
| Total de pacientes con trisomía 21 | n=9 | f |
|---|---|---|
| Cardiopatía congénita | ||
| Canal AV+/-PCA | 4 | 44% |
| CIV | 1 | 11% |
| CATVP | 1 | 11% |
| Estenosis mitral | 1 | 11% |
| PCA | 1 | 11% |
| PCA+CIA | 1 | 11% |
| Valores hemodinámicos | ||
| RVP/RVS | ||
| < 0,3 | 5 | 56% |
| 0,3 a 0,6 | 1 | 11% |
| > 0,6 | 3 | 33% |
| RVPi (UWood/m2) | ||
| <3 | 4 | 44% |
| 3 a 6 | 4 | 44% |
| >6 | 1 | 11% |
| PAPm (mm Hg) | ||
| 25-30 | 0 | 0% |
| 30-40 | 3 | 33% |
| >40 | 6 | 67% |
CANAL AV: canal atrioventricular, CIV: comunicación interventricular, CATVP: conexión anómala total de venas pulmonares, PCA: persistencia de conducto arterioso, CIA: comunicación interauricular.
Relación entre patrones hemodinámicos de RVPi y RVP/RVS, según el grado de PAP m
| RVPi (UW/m2) | n=57 | PAP m (mm Hg) | |||||
|---|---|---|---|---|---|---|---|
| 25-30 | 30-40 | >40 | |||||
| Total | 57 | 9 | 16% | 18 | 32% | 30 | 52% |
| <3 | 32 | 7(22%) | 11(34%) | 14(44%) | |||
| 3-6 | 16 | 2(13%) | 4(25%) | 10(63%) | |||
| >6 | 9 | 0 | 3(33%) | 6(67%) | |||
| RVP/RVS | 25-30 | 30-40 | >40 | ||||
| <0,3 | 31 | 7(23%) | 10(32%) | 14(45%) | |||
| 0,3-0,6 | 18 | 2(11%) | 7(39%) | 9(50%) | |||
| > 0,6 | 8 | 0 | 1(13%) | 7(88%) | |||
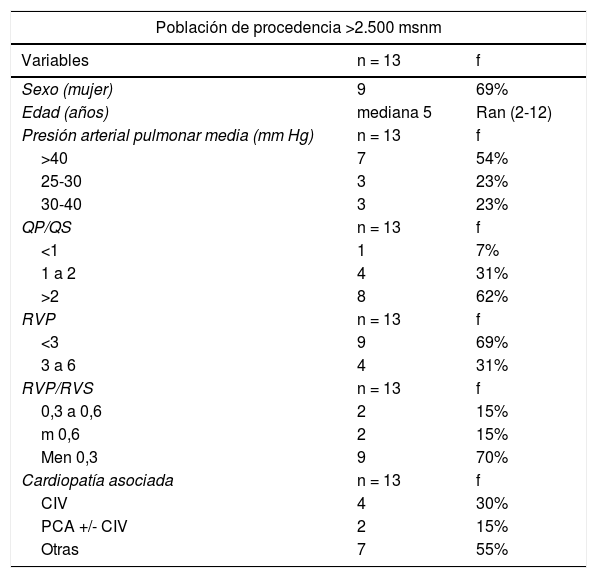
En relación con el estudio de vasorreactividad pulmonar (tabla 6), se observó que no en todos se realizaron las PVA, las cuales fueron positivas en 12 de 14 pacientes, y que la prueba más utilizada fue con O2 al 100% en 10 de 14 casos; en un paciente se utilizó iloprost. En la tabla 7 se detallan las características de la población de mediana a gran altura (> 2.500 msnm), es decir 13 de 57 pacientes. La cardiopatía más frecuente fue la comunicación interventricular seguida de la PCA, con valores altos de QP/QS y presión arterial pulmonar media y bajos de resistencias pulmonares.
Características de los pacientes de procedencia de > 2.500 msnm
| Población de procedencia >2.500 msnm | ||
|---|---|---|
| Variables | n = 13 | f |
| Sexo (mujer) | 9 | 69% |
| Edad (años) | mediana 5 | Ran (2-12) |
| Presión arterial pulmonar media (mm Hg) | n = 13 | f |
| >40 | 7 | 54% |
| 25-30 | 3 | 23% |
| 30-40 | 3 | 23% |
| QP/QS | n = 13 | f |
| <1 | 1 | 7% |
| 1 a 2 | 4 | 31% |
| >2 | 8 | 62% |
| RVP | n = 13 | f |
| <3 | 9 | 69% |
| 3 a 6 | 4 | 31% |
| RVP/RVS | n = 13 | f |
| 0,3 a 0,6 | 2 | 15% |
| m 0,6 | 2 | 15% |
| Men 0,3 | 9 | 70% |
| Cardiopatía asociada | n = 13 | f |
| CIV | 4 | 30% |
| PCA +/- CIV | 2 | 15% |
| Otras | 7 | 55% |
En cuanto a la epidemiología de la hipertensión pulmonar (tabla 1) en diferentes estudios se ha encontrado mayor frecuencia en mujeres en una relación de 1,8-2/1 no concordante con nuestros resultados13–15; sin embargo, cuando se estratifica por edad se encuentra que en el grupo de mayor frecuencia (1-3 años), la proporción mujer/varón es 2/1.
En distintas investigaciones no se ha logrado definir cuál es la edad en la que se desarrollará hipertensión pulmonar, porque depende a su vez de varios factores y de las enfermedades. En Bélgica existe un registro en el cual se encontró que en los pacientes con canal aurículo-ventricular, el síndrome de Eisenmenger era más común a una edad promedio de 11 años y por el contrario en la comunicación interauricular, la enfermedad vascular pulmonar se presentó después de los 20 a 30 años15. En nuestro medio no se cuenta con registros grandes ni estudios prospectivos para establecer estos datos. No obstante, es interesante el caso del paciente de menor edad (3.5 meses) con hipertensión pulmonar leve, a quien se le practicó cirugía de ventana aorto-pulmonar a los dos meses de edad, y además padecía comunicación interventricular tipo Gerbode, de 3mm, con la posibilidad de que la ventana aorto-pulmonar por sí sola no condicionara a la hipertensión pulmonar por la edad en la que se corrigió, sino por la presencia de los otros defectos16–18.
En cuanto al diagnóstico genético asociado según las guías19–22, la trisomía 21 se encuentra dentro del grupo asociado a mayor incidencia de hipertensión pulmonar, con o sin cardiopatías congénitas, hecho que concuerda con nuestro estudio, aunque también se halló un caso de síndrome de Turner, con diagnóstico de comunicación interventricular amplia e hipertensión pulmonar severa. No obstante, en las guías no se ha descrito este síndrome en asociación con hipertensión pulmonar, por lo que podría ser sólo un hallazgo; sin embargo debe observarse un mayor número de pacientes para aseverarlo. En cuanto a la sintomatología, gran parte tenía afectación de la clase funcional y de éstos la mayoría se encontraba en clase funcional II, dato que concuerda con el registro europeo, que reporta que el 51% se hallaba en clase funcional II (según la NYHA)23. Sin embargo, para determinar con mayor exactitud la clase funcional consideramos que ésta debería estratificarse según la clasificación de Panamá 2011, que depende del grupo etario y las actividades realizadas en cada uno, y permite valorarla de manera más fiable y cercana a la realidad, y aunque no estima la calidad de vida puede dar una idea de ella11,23.
Se utilizó la última clasificación de hipertensión pulmonar (tabla 2), donde el grupo 1fue el más frecuente; sin embargo, estos hallazgos no concuerdan completamente con el registro REVEAL23 que incluyó pacientes desde los 3 meses a 18 años de edad (al momento de ser enrolados), en el que predominaron las enfermedades del grupo 1 (92%), con mayor frecuencia de hipertensión arterial pulmonar idiopática y solo el 38% fueron cardiopatías congénitas. En nuestro estudio el subgrupo de cardiopatías congénitas fue el más frecuente, con menor frecuencia de hipertensión pulmonar idiopática/hereditaria (3 casos), hecho que puede obedecer a genotipos propios del país y a la accesibilidad del sistema de salud. Sin embargo, consideramos que esta nueva clasificación no se adapta por completo a la edad pediátrica ya que no refleja la compleja heterogeneidad de los factores de hipertensión pulmonar (anomalías cromosómicas, trastornos respiratorios del sueño, aspiración crónica y enfermedad pulmonar parenquimatosa secundaria que contribuyen a la enfermedad vascular pulmonar pediátrica). Así, se han propuesto otras clasificaciones que tienen en cuenta estos aspectos pero aún no son completamente validadas4,24.
Por otro lado, se ha subclasificado la hipertensión arterial pulmonar de las cardiopatías congénitas (Nice 2013), conocida como clasificación clínica2, que forma cuatro grupos; en la población del estudio predominó el grupo II (cortocircuito sistémico pulmonar). En Perú, el acceso a las cirugías cardiacas correctivas es limitado, lo que explicaría la diferencia con lo reportado por Zijlstra et al. en pacientes con hipertensión arterial pulmonar por cardiopatías congénitas, donde la mayor parte de sus casos (30%) se ubicó en el grupo IV (hipertensión arterial pulmonar posoperatoria), seguido de 24% en el grupo I (Eisenmenger) y en menor frecuencia del grupo II. Asimismo, encontró que el 11% de sus pacientes no pudieron clasificarse y propuso que debería añadirse un grupo V (categoría no clasificable), lo cual también recomendamos; aunque los estudios con esta subclasificación son escasos, ésta no puede abarcar toda la heterogeneidad y el amplio espectro de cardiopatías congénitas, por ejemplo en la transición entre el grupo I (complejo Eisenmmenger) y el II (secundario a cortocircuito sistémico pulmonar), pueden existir pacientes que estén en el límite de ambos, siendo poco factible la unificación de criterios que los definan adecuadamente6,25.
Las cardiopatías congénitas fueron las enfermedades cardiacas más asociadas a hipertensión pulmonar, por ello en este estudio se considera importante resaltar el antecedente de cirugía correctiva o paliativa, pues se encontró que el 78,9% de los casos no había sido sometido a ninguna cirugía cardiaca previa y que solo el 8,8% había sido intervenido previamente mediante cirugía correctiva, lo cual es parte de nuestra problemática de salud pública ya que los centros especializados en estas cirugías cardiacas son escasos y se concentran en la capital, forma en la que en algunos se ha optado por realizar cirugías paliativas como “banding pulmonar”. No obstante, esta situación no es ajena a los países más desarrollados, como lo demuestra el mayor registro europeo de hipertensión pulmonar (REVEAL), que encontró que los defectos no fueron reparados o fueron reparados parcialmente en el 62,3%; de la misma forma Zijlstra6 reporta que el 62% de sus casos no fueron corregidos23.
Los niños con trisomía 21 constituyen una población específica cuyos parámetros hemodinámicos no están bien estudiados. En nuestro estudio se halló que la cardiopatía congénita más frecuente asociada a hipertensión arterial pulmonar en estos pacientes fue el canal AV con o sin PCA (tabla 4); similar a otros estudios19,20,23,26, respecto a la hemodinamia, la mayor parte de pacientes tenían valores altos de presión arterial pulmonar media, pero con resistencias pulmonares bajas, es decir, no había compromiso de la vasculatura pulmonar. Aunque no existen muchos estudios con los que se puedan comparar nuestros resultados y nuestro número de casos fue 9, se encontró que algunas investigaciones han propuesto que los cambios de la vasculatura pulmonar se presentan en forma más temprana y rápidamente más progresiva en pacientes con trisomía 21 a diferencia de niños con cromosomas normales y cardiopatías congénitas similares19,20. Sin embargo, otros autores20-22,27 indican que si bien los niños con síndrome de Down tienen una alta incidencia de hipertensión arterial pulmonar (hipertensión arterial pulmonar), esta hipertensión no es más severa que la observada en pacientes sin este síndrome.
Con relación a los parámetros hemodinámicos de toda la población estudiada, aproximadamente la mitad tuvo valores que consideramos altos, como era de esperarse, porque una de las principales indicaciones de cateterismo es la confirmación de presión pulmomar obtenida por ecocardiografía, y en menor frecuencia se encontraron valores considerados leves (25-30mm Hg); no obstante, éstos pueden estar influenciados por factores ya descritos como la sedación durante el acto intervencionista24,25.
Los valores de resistencia pulmonar definen la decisión terapéutica, es decir, en caso de cardiopatías congénitas cuando se encuentran valores bajos, está indicada la cirugía correctiva; afortunadamente en nuestro estudio las RVP altas (RVPi > 6 UW/m2 y RVP/ RVS > 0,6) fueron las menos frecuentes y esto se reflejó en la conducta terapéutica, ya que en el 52% de los casos se reparó el defecto, lo que fue posible dado que cursaban con resistencias bajas. No se encontró una relación directamente proporcional entre la presión arterial pulmonar media y las RVPi y RVP/RVS, dado que tener presión arterial pulmonar media > 40mm Hg, no necesariamente implica incremento de la resistencia pulmonar, y al observar el grupo que presentó valores de presión arterial pulmonar media moderadas (30-40mm Hg), éste ya tenía valores altos de resistencia pulmonar, por lo tanto, presiones pulmonares muy elevadas no reflejan la gravedad de la hipertensión pulmonar, o daño en la vasculatura pulmonar y aún no hay ningún estudio que pueda establecer el punto donde se empiezan a incrementar las resistencias pulmonares. Por tanto, proponemos que para definir el grado de severidad de hipertensión pulmonar se tengan en cuenta los valores antes mencionados de RVPi y RVP/RVS que concuerda mejor con el grado de afectación anatomo-fisiológico23,28,29.
En nuestro instituto no todos los pacientes fueron sometidos a PVA, ya que como se ha descrito anteriormente la indicación fue el hallazgo de resistencia pulmonar alta, el iloprost, que se menciona en las guías; no es de fácil acceso en nuestro país. Las PVA con oxígeno en niños con cardiopatías congénitas han sido criticadas porque el consumo de oxígeno no se puede medir simultáneamente y la diferencia arteriovenosa de oxígeno se hace muy pequeña, lo cual aumenta los errores en los cálculos hemodinámicos23,25. El 85% de nuestros niños fueron respondedores positivos, proporción que es más alta que en otros estudios; no obstante se requieren estudios dado que sólo se realizaron 14 PVA. De otra parte, Barts23 encontró que el 27% de sus casos respondían de forma positiva, mientras que otros registros en las diferentes regiones europeas muestran porcentajes muy variables, desde menos del 10% hasta 50%; sin embargo, hay controversia sobre la frecuencia de los respondedores positivos en pediatría. Otra prueba realizada fue la oclusión temporal del defecto mediante balón, presentando descenso de la PAP que indicó la idoneidad para oclusión definitiva del defecto, hecho que concuerda con otros estudios23,28,29.
Aunque pocos estudios informan sobre la influencia de la altura, es un parámetro importante poco estudiado en hipertensión pulmonar pediátrica. Por su parte, Dalen reporta que la incidencia de hipertensión pulmonar puede ser mayor en niños con comunicación interauricular que residen entre 1.300 y 1.600 msnm24,26. La altura es un factor importante en la biopatogénesis de la hipertensión pulmonar, hay tres factores básicos: la hipoxia hipobárica, el remodelamiento vascular pulmonar retardado y la reactividad del lecho vascular pulmonar30; otro aspecto es la producción disminuida del óxido nítrico31. En nuestro estudio se encontró (tabla 7) que el 22,8% era procedente de moderada altura (> 2.500 msnm) y que la cardiopatía más frecuente asociada a hipertensión arterial pulmonar fue la comunicación interventricular. Este resultado no fue lo esperado, ya que hubiera sido más congruente encontrar PCA, porque ésta es la cardiopatía más frecuente en mediana a grandes alturas, probablemente existen factores genéticos propios de población peruana, que lo pueda explicar, pero cuando evaluamos los parámetros hemodinámicos, la presión arterial pulmonar media más frecuente fue > 40mm Hg con RVP leves, cursando con el mismo comportamiento hemodinámico que el resto de la población de estudio. Por otra parte, la hipertensión pulmonar secundaria a cardiopatías congénitas aparece más temprano, en relación con los factores ya mencionados, pero en nuestro estudio la edad más frecuente fue 5 años, un poco más tardía que en la población general de estudio (3 años), resultados que podrían estar influenciados por el número de pacientes. Por consiguiente, recomendamos ampliar estudios con mayor número pacientes para conocer el comportamiento de la hipertensión pulmonar en esta población específica.
ConclusiónLa hipertensión pulmonar pediátrica tiene características propias; el principal grupo lo constituye la hipertensión arterial pulmonar (grupo 1), siendo importante la asociación a cardiopatías congénitas. La resistencia vascular pulmonar puede estar incrementada aún en estadios clínicamente no significativos, es por ello que el diagnóstico y tratamiento oportuno (quirúrgico correctivo o médico) es esencial para evitar su progresión y el daño irreversible de la vasculatura pulmonar, y en el futuro cuando estos pacientes sean una población adulta evitar las repercusiones negativas en todos los aspectos, aún en el socioeconómico. Este estudio constituye el primer registro de hipertensión pulmonar (por cateterismo cardiaco) en niños peruanos; sin embargo, se requieren estudios complementarios prospectivos que realicen el seguimiento a largo plazo de estos pacientes, sin olvidar algunos grupos específicos (según procedencia y alteraciones cromosómicas), para definir pronóstico, evolución y tratamiento de la hipertensión pulmonar pediátrica en Perú.
Conflictos de interésNinguno.
FinanciaciónAutofinanciado.