



La anemia de células falciformes es una hemoglobinopatía hereditaria producida por la presencia de hemoglobinaS, que disminuye la solubilidad y a través del proceso de polimerización da lugar a hematíes en forma de hoz que obstruyen la red microvascular. Se caracteriza por episodios de daño por isquemia-reperfusión que contribuyen a la disfunción de órganos vitales. El advenimiento de la terapia inductora de hemoglobina fetal, asociada a la profilaxis antibiótica, ha permitido prolongar la supervivencia. Sin embargo, al incrementar la longevidad, las complicaciones cardiovasculares se hacen evidentes con el desarrollo de vasculopatía sistémica, infarto agudo de miocardio, hipertensión arterial pulmonar y disfunción ventricular. El objetivo de este artículo es revisar la fisiopatología y el tratamiento de las principales manifestaciones cardiovasculares en pacientes con anemia de células falciformes.
Sickle cell anemia is an inherited hemoglobinopathy caused by the presence of hemoglobinS, which lowers the solubility and through the process of polymerization results in sickle erythrocytes obstructing the microvascular network. This entity is characterized by episodes of ischemia-reperfusion injury to vital organs, contributing to its dysfunction. The advent of inducing fetal hemoglobin therapy associated with antibiotic prophylaxis has allowed prolonged survival. However, by increasing longevity, cardiovascular complications are evident, with the development of systemic vascular disease, acute myocardial infarction, pulmonary arterial hypertension and left ventricular dysfunction. The aim of this paper is to review the pathophysiology and treatment of major cardiovascular events in patients with sickle cell anemia.
Se han identificado múltiples formas de hemoglobinopatías estructurales relacionadas con alteraciones de la cadena beta, dentro de las cuales la más relevante es la drepanocitosis o anemia de células falciformes.
Es una enfermedad autosómica que sigue un patrón mendeliano común, cuya alteración resulta de la sustitución del aminoácido ácido glutámico por valina en la posición 6 de la cadena beta, dando lugar a una hemoglobina falciforme (HbS) mutante.
Es de gran prevalencia en personas de raza negra y su mestizaje, los cuales pueden ser portadores de hemoglobinaS en su forma homocigota (HbSHbS) o heterocigota (HbAHbS); en este último caso da lugar solo al rasgo falciforme1.
La HbS funciona de manera normal excepto cuando es desoxigenada, ya que conduce a su polimerización patológica. Este proceso conlleva la formación de una red gelatinosa de polímeros fibrosos responsables de la deformación del hematíe en forma de hoz, que rigidizan su membrana, disminuyen su flexibilidad y aumentan la viscosidad sanguínea2. Como consecuencia, los hematíes no pueden atravesar con éxito los pequeños capilares, de ahí que provocan episodios de oclusión microvascular y prematura destrucción de los mismos, lo que conduce a anemia hemolítica crónica grave3.
La polimerización de la hemoglobina es el mecanismo mejor estudiado al que se le atribuye la principal complicación de esta enfermedad, daño por isquemia-reperfusión, que conduce finalmente a infarto de órganos diana, entre ellos bazo, riñones, hígado, músculo, cerebro, pulmón y hueso4,5. La complicación más común es la crisis vasooclusiva típica que se da como resultado del proceso de necrosis de la médula ósea6.
Mucho antes del advenimiento de la hidroxiurea y la realización de transfusiones profilácticas en niños, se llevó a cabo el Cooperative Study of patients with Sickle Cell Disease (CSSCD, por su sigla en inglés), un gran estudio que puso en evidencia la pobre expectativa de vida para estos pacientes, indicando una edad media de vida de 48 y 42años para mujeres y hombres, respectivamente7.
En la actualidad este panorama ha cambiado de forma considerable y la expectativa de vida se ha incrementado de forma ostensible3, hecho que se explica por la mejoría en la disponibilidad y seguridad de las transfusiones sanguíneas, sumado a la terapia inductora de hemoglobina fetal con hidroxiurea y al uso de penicilina profiláctica, con lo cual se redujeron significativamente las tasas de infección y sus complicaciones8.
Sin embargo, este hecho ha puesto en evidencia múltiples comorbilidades y complicaciones en los pacientes, que al llegar a la edad adulta y/o la vejez son víctimas de lesiones crónicas de órganos blanco, consecuencia de un proceso de daño vascular sistémico y pulmonar progresivo3,9. Esta revisión resume la evidencia actual disponible respecto a las complicaciones cardiovasculares de mayor relevancia en el paciente con anemia de células falciformes, como infarto agudo de miocardio, hipertensión arterial pulmonar y disfunción ventricular, en vista de su gravedad e impacto en la mortalidad.
Infarto agudo de miocardioBien se sabe que el infarto agudo de miocardio se asocia comúnmente con enfermedad coronaria aterosclerótica, y que más del 80% de estos se atribuyen a rotura de una placa vulnerable10.
El hecho de que la mayoría de pacientes con anemia de células falciformes sean jóvenes sin factores de riesgo, con frecuentes crisis vasooclusivas que se presentan como episodios de dolor corporal difuso o dolor torácico atípico, enmascara la entidad. A ello se suma que los cambios electrocardiográficos puedan ser inespecíficos, con poco valor diagnóstico, lo cual hace que el infarto agudo de miocardio en anemia de células falciformes sea subdiagnosticado y tenga alta mortalidad por falta de evidencia en modalidades terapéuticas11.
PatogénesisEl mecanismo exacto que explique el infarto de miocardio aún se desconoce, pero se cree que es consecuencia de la conjugación de múltiples factores, entre ellos la anemia, los efectos reológicos y morfobioquímicos de las células falciformes y la anomalía funcional plaquetaria12.
La anemia y la hipoxia secundaria conducen a isquemia miocárdica por reducción en la oxigenación, aunque también por anormalidades en la microvasculatura miocárdica, específicamente la displasia fibromuscular de los pequeños vasos coronarios presente en pacientes con anemia de células falciformes13.
El proceso de producción de células falciformes lleva a oclusión vascular, hipoxia tisular subsecuente y daño por reperfusión, que conducen a inflamación y daño endotelial, proceso que se ve agravado por la tendencia de las células falciformes a adherirse al endotelio en sitios de alto flujo y turbulencia. Dichos factores reológicos de viscosidad alterada, poca flexibilidad y aglomeración de hematíes causan mayor obstrucción vascular coronaria, isquemia e infarto14. Adicionalmente, la lesión mecánica crónica a las células endoteliales debida a la rigidez eritrocitaria puede desencadenar un ataque autoinmunológico endotelial y perpetuar un círculo vicioso de daño15.
Cuando se produce injuria por isquemia-reperfusión se activa la enzima xantina oxidasa, que conduce a la producción de radicales libres, los cuales estimulan el proceso de estrés oxidativo en las células endoteliales y al mismo tiempo inducen expresión de moléculas de adhesión, como moléculas de adhesión vascular (VCAM), moléculas de adhesión intercelular (ICAM), selectina-P y selectina-E16.
De otro lado, la activación plaquetaria libera ADP, moléculas adhesivas, como selectina-P, trombospondina, fibrinógeno y factor de Von Willebrand, así como factores de coagulación y crecimiento17,18.
En pacientes con anemia de células falciformes se han encontrado niveles circulantes de diversos marcadores de activación plaquetaria. Es así como el tromboxano liberado de las plaquetas, desempeña un papel importante al inducir al vasoespasmo, fenómeno que induce un fenotipo inflamatorio y de mayor adhesión de hematíes falciformes al endotelio19.
Por otra parte, la elevación de los niveles de homocisteína se considera como un factor de riesgo bien establecido para trombosis venosa y arterioesclerosis. De igual forma, el riesgo cardiovascular incrementado asociado a la hiperhomocisteinemia se ha relacionado estrechamente con la disfunción de las células endoteliales inducida por la homocisteína, al tiempo que esta ejerce una acción tóxica directa en los vasos sanguíneos20.
Uno de los mecanismos mejor estudiados es la interferencia que tiene la homocisteína con la síntesis de endotelina-1, péptido vasoactivo sintetizado por las mismas células endoteliales que, además de regular el tono vasomotor, limita la activación inflamatoria y mantiene a la superficie endotelial en estado no trombótico. También se asocia con el incremento del factor de crecimiento endotelial vascular (VEGF), estimula la proliferación de las células del músculo liso vascular y promueve la oxidación del colesterol LDL debido a la presencia de sus compuestos sulfhidrilo, contribuyendo de esta forma al desarrollo de aterosclerosis20.
Se han encontrado concentraciones plasmáticas de homocisteína elevadas en pacientes con anemia de células falciformes21, y aun mucho más altas si tenían historia de episodio isquémico cerebral comparado con aquellos sin este antecedente.
La causa de la hiperhomocisteinemia no es clara. Algunos autores creen que puede estar relacionada con deficiencia de ácido fólico. De hecho, Brattstrom et al.22 hace más de 2 décadas lograron demostrar que el ácido fólico tenía un efecto reductor importante en los niveles de homocisteína, ya que actúa induciendo su re-metilación a metionina, y en ese momento se planteó el tratamiento con ácido fólico como medida profiláctica para evitar la hiperhomocisteinemia.
DiagnósticoDada su presentación atípica, se requiere un alto índice de sospecha clínica. Usualmente los pacientes tienen pocos o ningún factor de riesgo para enfermedad coronaria, por tanto las herramientas de estratificación de riesgo, como las escalas de Global Registry of Acute Coronary Events (GRACE, su sigla en inglés) y Thrombolysis in Myocardial Infarction (TIMI, su sigla en inglés), no son útiles y los clasifican como pacientes de bajo riesgo11.
El uso de electrocardiograma es de poca utilidad pues los cambios inespecíficos de ST-T y los signos de repolarización anormal son comunes en estos pacientes23. El papel de las enzimas cardiacas es bastante cuestionable si se tiene en cuenta que el mecanismo propuesto para la muerte de los miocitos en anemia de células falciformes ocurre por apoptosis en lugar de necrosis24, y en consecuencia no hay elevación de enzimas cardiacas. Además, los marcadores séricos para daño miocárdico, incluyendo la troponinaT, se pueden elevar durante las crisis dolorosas como consecuencia del daño esquelético, haciéndolos inespecíficos12.
En la actualidad se carece de estudios a gran escala que puedan validar a la troponinai (TnI) como marcador fiable de infarto de miocardio en pacientes con anemia de células falciformes. En el estudio de Aslam et al.25, diseñado para evaluar el comportamiento de la TnI durante las crisis falciformes, participaron 32 pacientes, todos con cuadro clínico inicial de crisis dolorosas. Los síntomas tenían por lo menos un día de evolución con el fin de aumentar la sensibilidad de los hallazgos anormales en los niveles de TnI, considerando como elevados niveles mayores o iguales a 0,4ng/ml.
De 32 pacientes, solo 2 tuvieron TnI elevado, cuya característica común fue el síndrome torácico agudo a su ingreso, lo que sugirió considerar a la TnI como marcador de isquemia miocárdica durante las crisis vasooclusivas25.
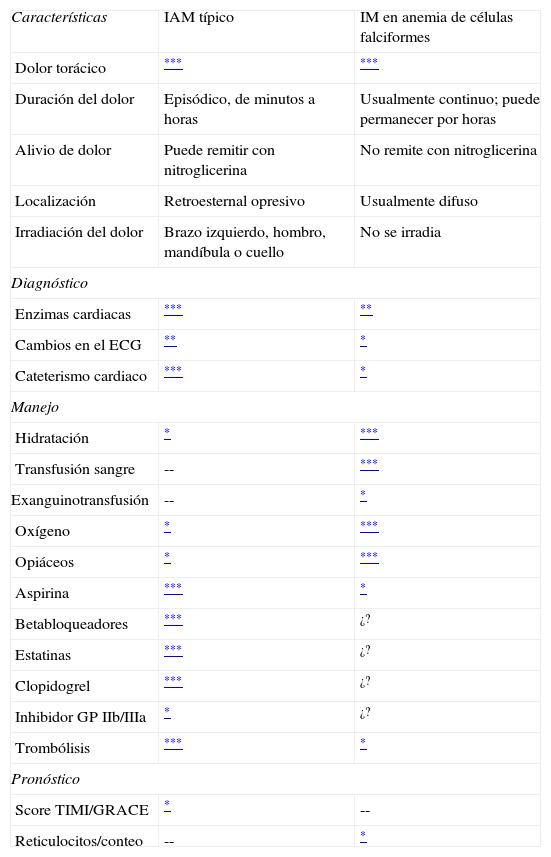
Puede concluirse, entonces, que la elevación de los niveles de troponinai, a pesar de sus limitaciones y su inespecificidad en este contexto, sumada a criterios clínicos, puede ser útil en el diagnóstico de infarto de miocardio. Las características básicas del infarto agudo de miocardio en anemia de células falciformes se resumen en la tabla 1.
Características de infarto agudo de miocardio típico comparado con infarto agudo de miocardio en anemia de células falciformes
| Características | IAM típico | IM en anemia de células falciformes |
| Dolor torácico | *** | *** |
| Duración del dolor | Episódico, de minutos a horas | Usualmente continuo; puede permanecer por horas |
| Alivio de dolor | Puede remitir con nitroglicerina | No remite con nitroglicerina |
| Localización | Retroesternal opresivo | Usualmente difuso |
| Irradiación del dolor | Brazo izquierdo, hombro, mandíbula o cuello | No se irradia |
| Diagnóstico | ||
| Enzimas cardiacas | *** | ** |
| Cambios en el ECG | ** | * |
| Cateterismo cardiaco | *** | * |
| Manejo | ||
| Hidratación | * | *** |
| Transfusión sangre | -- | *** |
| Exanguinotransfusión | -- | * |
| Oxígeno | * | *** |
| Opiáceos | * | *** |
| Aspirina | *** | * |
| Betabloqueadores | *** | ¿? |
| Estatinas | *** | ¿? |
| Clopidogrel | *** | ¿? |
| Inhibidor GP IIb/IIIa | * | ¿? |
| Trombólisis | *** | * |
| Pronóstico | ||
| Score TIMI/GRACE | * | -- |
| Reticulocitos/conteo | -- | * |
ECG: electrocardiograma: IAM: infarto agudo de miocardio.
Adaptada de Pannu et al.11.
¿? Hay falta de consenso/evidencia inconsistente respecto a su utilidad.
Pueden considerarse otras modalidades diagnósticas, como el ecocardiograma, que evidencia anormalidades en la motilidad de la pared cardiaca, al tiempo que permite evaluar el rendimiento cardiaco. Sin embargo, no hay reportes de su utilidad en el diagnóstico de infarto agudo de miocardio en pacientes con anemia de células falciformes11.
La resonancia magnética cardiaca tiene la ventaja de proporcionar la caracterización no invasiva del miocardio en alta resolución espacial. Por su parte, la coronariografía tiene un papel muy limitado, suele ser normal y a menudo no existe enfermedad coronaria epicárdica que sea susceptible a revascularización mecánica; por tanto, no se recomienda como procedimiento de rutina en pacientes jóvenes con anemia de células falciformes y perfiles de riesgo bajo12.
ManejoLa falta de reconocimiento de esta entidad, aunada a la deficiencia de guías o protocolos establecidos de tratamiento, hace que existan diversas modalidades terapéuticas.
La deshidratación y desoxigenación contribuyen a incrementar la formación de células falciformes y empeoran la vasooclusión; por esto, la hidratación y oxigenación adecuadas constituyen el pilar básico en la terapia de manejo. Por supuesto, debe haber un buen control del dolor, ya que en etapas agudas aumenta la respuesta simpática y la carga miocárdica de oxígeno por incremento en su demanda.
La transfusión sanguínea mejora la oxigenación en estos pacientes26. La meta es incrementar el hematócrito a 30% o mantener niveles de hemoglobina a 11g/dl27,28, en los cuales es menos probable la formación de hematíes falciformes y por ende hay mejor oxigenación.
La exanguinotransfusión se recomienda en pacientes con infarto de miocardio severo y progresivo, en cuyos casos la meta es disminuir el nivel de HbS a menos de un 30% sin exceder a un nivel de Hb de 10g/dl27,28. Con esta terapia se han reportado resultados positivos, y supervivencia de todos los pacientes sometidos a esta29,30 al reemplazar directamente hematíes portadores de HbS por otros, con HbA normal. Así se logra reducir la viscosidad en la microcirculación y los marcadores inflamatorios (glóbulos blancos, plaquetas, moléculas de adhesión VCAM-1), lo cual es suficiente para contrarrestar el inicio de la cascada trombótica31.
En la actualidad, las demás intervenciones terapéuticas recomendadas para infarto agudo de miocardio típico no tienen evidencia que avale su uso en el contexto de esta enfermedad.
Greenberg et al.32 evaluaron el efecto de bajas dosis de aspirina en frecuencia y severidad de crisis dolorosas en niños sin hallar beneficios. Aún no hay reporte de estudios que evalúen el uso de clopidogrel ni de inhibidores de la glucoproteínaIIb/IIIa.
El tratamiento a largo plazo debe dirigirse a la prevención de recurrencia de infarto agudo de miocardio, lo cual incluye disminución en el riesgo de infección con profilaxis antibiótica e inmunización, así como disminución en la formación de hematíes falciformes con hidroxiurea y terapia transfusional crónica. La hidroxiurea sigue siendo el pilar terapéutico, ya que induce la síntesis de hemoglobina fetal, la cual inhibe el proceso de polimerización de HbS y adicionalmente reduce la adhesión de reticulocitos al endotelio vascular, modula el proceso inflamatorio e induce síntesis de ON33.
Hipertensión arterial pulmonarLa hipertensión pulmonar representa una de las principales complicaciones emergentes y cada vez es más evidente en esta población de adultos mayores; adicionalmente, se considera como un factor de riesgo independiente para muerte en la población con anemia de células falciformes.
En la hipertensión pulmonar se elevan las presiones dentro del sistema pulmonar, fenómeno que se caracteriza por disfunción endotelial y desequilibrio entre vasodilatadores-vasoconstrictores, en asociación con hiperplasia progresiva del músculo liso y neointimal con obliteración subsecuente de la circulación arteriolar34. Este incremento progresivo de la resistencia vascular pulmonar lleva a disfunción ventricular derecha y reducción del gasto cardiaco.
PatogénesisEl bazo es el sitio natural por donde se eliminan glóbulos rojos con algún grado de anormalidad. En pacientes con anemia de células falciformes este órgano es sometido a vasooclusiones e infartos recurrentes, lo cual conlleva autoesplenectomía. La asplenia funcional perpetúa e incrementa las tasas de hemólisis intravascular, esta última establecida como el principal mecanismo responsable de la vasculopatía pulmonar crónica35,36.
La severidad de la hemólisis intravascular medida rutinariamente de forma indirecta a través de ciertas variables, entre ellas recuento de reticulocitos, niveles bajos de haptoglobina, niveles altos de bilirrubina total, aspartato aminotransferasa y lactato deshidrogenasa, se asocia con incremento de complicaciones vasculares37.
Disfunción endotelialEl motivo por el cual la anemia hemolítica se propone como protagonista en la alteración de la integridad y funcionalidad del lecho vascular obedece a que es la responsable de la inhibición de la síntesis de ON38, el cual promueve la vasodilatación arterial y mantiene la homeostasis vascular al disminuir la expresión endotelial de moléculas de adhesión. Así mismo, disminuye la activación plaquetaria e inhibe la mitogénesis y proliferación de fibroblastos, así como de células musculares lisas y endoteliales.
La hemólisis produce liberación de hemoglobina en plasma, la misma que se encarga de neutralizar el ON al oxidarlo a nitrato. Niveles discretos de hemoglobina libre en plasma son suficientes para inhibir toda la señalización de ON39. Por otro lado, la hemólisis libera una enzima de los hematíes, la arginasa1, que lleva a cabo la hidrólisis de arginina a ornitina, reduciendo el sustrato para la síntesis de ON40.
Los eritrocitos falciformes incrementan la producción de endotelina-1 y factor de crecimientoB derivado de plaquetas, ambos con propiedades mitogénicas y quimiotáxicas41. En estados hipóxicos crónicos se liberan de igual forma factor de crecimiento derivado del endotelio vascular y trombospondina-1, factores que contribuyen sustancialmente al desarrollo de la vasculopatía pulmonar falciforme, caracterizada no solo por vasoconstricción, sino por una remodelación estructural dada por hiperplasia intimal-muscular y trombosis in situ42.
Con base en lo anterior se puede concluir que, desde el punto de vista fisiopatológico, existen 2 mecanismos principales que dan lugar a 2 fenotipos clínicos en el contexto de complicaciones crónicas: la disfunción endotelial inducida por hemólisis, que desencadena una vasculopatía secundaria con desarrollo no solo de hipertensión pulmonar, sino también de úlcera cutánea en extremidades inferiores, proteinuria, insuficiencia renal, hipertensión arterial sistólica y accidente cerebrovascular, y otras complicaciones relacionadas con crisis vasooclusivas y síndrome torácico agudo, en las que predominan los niveles estables de hemoglobina (menos hemólisis y mayor viscosidad), recuento elevado de leucocitos (patrón inflamatorio) y niveles bajos de hemoglobina fetal con tasas altas de polimerización43.
Estos mecanismos no son excluyentes mutuamente; al contrario, en muchos pacientes hay superposición de estos. Durante una crisis vasooclusiva pueden ocurrir fenómenos de hiperhemólisis que conducen a la activación de ambos mecanismos. Es así como podría explicarse la elevación de las presiones pulmonares durante el curso de un síndrome torácico agudo y posterior desarrollo de falla cardiaca derecha aguda44.
DiagnósticoNumerosos estudios de cohorte han dejado en evidencia la asociación entre la severidad de la anemia hemolítica con incrementos en la presión sistólica de la arteria pulmonar (PsAP) estimada por ecocardiografía y el alto riesgo de mortalidad en estos pacientes35,45-47.
El análisis CSSCD7 revela que los niveles elevados de propéptido natriurético tipo B N-terminal (NT-proBNP) por encima de 160pg/ml están estrechamente relacionados con la hipertensión pulmonar. Es el mayor predictor independiente de mortalidad (RR: 6,24, intervalo de confianza al 95% [IC95%]: 2,9-13,3, p<0,0001), estableciéndose como un biomarcador para hipertensión arterial pulmonar en pacientes con anemia de células falciformes47.
De igual manera, niveles altos de NT-proBNP se asociaron con los marcadores de anemia hemolítica, entre ellos niveles bajos de hemoglobina (p<0,001), así como niveles altos de lactato deshidrogenasa (p<0,001) y bilirrubina total (p<0,007)48.
Cribado para hipertensión arterial pulmonarEl ecocardiograma es un método no invasivo que permite realizar un cálculo estimado de la PsAP a partir de la medición de la velocidad de regurgitación tricúspide (VRT) en pacientes con anemia de células falciformes. Estas estimaciones ecocardiográficas han demostrado ser fiables y tener una buena correlación con la prueba diagnóstica definitiva, el cateterismo cardiaco derecho45.
Se han desarrollado 3 grandes estudios de cribado: National Institutes of Health (NIH)-PH cohort45, Duke cohort46 y University of North Carolina Chapel Hill cohort46. A partir de sus estimaciones, se valoró una VRT de 2,5m/s o más como punto de corte para pacientes de alto riesgo, y valores iguales o mayores de 3,0m/s como punto de corte para el diagnóstico de hipertensión pulmonar.
Desde esta perspectiva, los datos sugieren que valores de VRT menores a 2,5m/s o un NT-proBNP menor a 160pg/ml son normales y se asocian a bajo riesgo de muerte. Por el contrario, pacientes con VRT mayor a 3m/s tienen un riesgo relativo de muerte de 10,6 (IC95%: 3,3-36,6, p<0,001), en cuyos casos es obligatorio el cateterismo cardiaco derecho4.
En tal sentido, existe gran debate y controversia respecto al denominado grupo intermedio o borderline, con valores de VRT entre 2,5 y 2,9m/s. En este grupo de pacientes existe riesgo de muerte de 4,4 (IC95%: 01,06-12,02, p<0,001). A pesar de que solo el 25% del grupo intermedio tendrá hipertensión pulmonar confirmada por cateterización derecha, existe riesgo alto de progresión a hipertensión pulmonar franca y muerte34.
Las guías publicadas recientemente por la American Thoracic Society y aprobadas por la Pulmonary Hypertension Association y el American College of Chest Physicians49 establecen entre sus recomendaciones que todos los pacientes estables deben ser tamizados con ecocardiograma o NT-proBNP cada uno a 3 años para estatificación del riesgo. En casos de VRT mayor o igual a 2,5m/s y/o NT-proBNP mayor a 160pg/mL, se sugiere alto riesgo de desarrollar hipertensión pulmonar y elevada mortalidad, por tanto, a estos pacientes deben practicárseles coronariografía derecha para confirmar el diagnóstico y establecer estrategias terapéuticas. Por supuesto, la indicación de cateterismo derecho en pacientes con VRT mayor o igual a 3m/s es indiscutible.
Diagnóstico de cateterismo cardiaco derechoEl cateterismo cardiaco derecho es el estándar de oro para el diagnóstico de hipertensión pulmonar, cuya definición establecida por consenso es una presión media de la arteria pulmonar (mPAP) mayor o igual a 25mmHg. En la tabla 2 se resumen los datos relevantes de 3 nuevos estudios en pacientes con anemia de células falciformes que cursan con hipertensión arterial pulmonar diagnosticada a través de cateterismo cardiaco50-52.
Estudios de cateterismo cardiaco derecho en anemia de células falciformes
| Primer autor (Ref. #) | Mehari et al.51 | Parent et al.50 | Fonseca et al.52 |
|---|---|---|---|
| Número de tamizajes | 533 | 445 | 80 |
| Exclusiones | Ninguna | Insuficiencia renal, enfermedad hepática, enfermedad pulmonar restrictiva | Ninguna |
| Número de CCD | 86 | 96 | 26 |
| Sujetos con HP (mPAP≥25mmHg); % de CCD con HP | 56; 65% | 24; 25% | 8; 31% |
| mPAP (mm Hg) | 36±9 | 30±6 | 33±9 |
| Mortalidad del grupo de HP | 36% | 13% | 38% |
ACF: anemia de células falciformes; CCD: cateterismo cardiaco derecho; HP: hipertensión pulmonar; mPAP: presión media de arteria pulmonar.
Adaptada de Gladwin y Sachdev42.
El primer estudio, de Mehari et al.51, evidencia el alto riesgo de muerte incluso con elevaciones moderadas de mPAP (36mmHg). Tras un seguimiento de 4,4años se confirmó que la mortalidad fue significativamente más alta en el grupo con hipertensión arterial pulmonar comparada con la del grupo cuyas estimaciones ecocardiográficas de PsAP fueron normales (36% vs. 13%).
En el segundo estudio, hecho por Parent et al.50, se encontró que el 25% de pacientes con VRT mayor a 2,5m/s tenían mPAP≥25mmHg, además de una fuerte asociación entre hipertensión pulmonar, disminución de la capacidad funcional y mayor riesgo de muerte en comparación con el grupo hipertensión pulmonar vs. grupo de no hipertensión pulmonar, 12,5% vs. 0,3%, respectivamente. De igual manera se evidencian una vez más los factores de riesgo más importantes para hipertensión pulmonar, los marcadores de hemólisis y colestasis hepática, así como la enfermedad renal.
Es confuso y genera controversia el hecho de haber utilizado los criterios de exclusión expuestos en la tabla 2, ya que son precisamente esas complicaciones los factores de riesgo más significativos para hipertensión arterial pulmonar en pacientes con anemia de células falciformes45,53,54. Esto está fundamentado en el estudio multicéntrico Walk- treatment of Pulmonary Hypertension and Sickle Cell Disease with sildenafil Therapy (Walk-PHASST, por su sigla en inglés), en el que se mostró que el 6,4% de pacientes con HbS tenían un TFG menor a 30ml/min, y de estos el 91,7% tenía una VRT mayor o igual a 2,5m/s y el 13% mayor o igual a 3,0m/s, valores que indican que la prevalencia de hipertensión pulmonar en los pacientes con enfermedad renal es alta53.
TratamientoHoy día no existen grandes ensayos clínicos aleatorizados que establezcan recomendaciones de alta calidad para dirigir una terapia específica. Durante los episodios de crisis vasooclusivas, síndrome coronario agudo o falla ventricular derecha se puede desarrollar hipertensión arterial pulmonar, motivo por el cual se recomienda el control de la enfermedad subyacente y la utilización de hidroxiurea. Esta última continúa siendo la piedra angular terapéutica, pues su finalidad es reducir la severidad de la anemia hemolítica al aumentar la concentración de hemoglobina fetal. Así mismo, se indican las transfusiones sanguíneas, ya que mejoran sustancialmente la oxigenación tisular.
La citoquina inflamatoria endotelina-1 (ET-1) es conocida como promotora de vasoconstricción, fibrosis y proliferación celular al unirse a los receptores ET-A y ET-B, de igual forma implicada en la aceleración de la hemólisis eritrocitaria55.
En una pequeña serie de casos y controles de 17 pacientes con anemia de células falciformes e hipertensión arterial pulmonar se inició tratamiento con bosentán (bloqueador de receptores de endotelinaA y B) o ambrisentán (bloqueador selectivo de receptor de endotelinaA), dando como resultado disminución de los niveles de NT-proBNP y en las medidas de VRT, lo que indicaría mejoría de la hipertensión arterial pulmonar56. Sin embargo, debido al menor riesgo de disfunción hepatocelular con ambrisentán, este se prefiere como agente de primera línea.
La terapia con inhibidores de la fosfodiesterasa5 fue inicialmente promisoria. El estudio Walk-PHaSST53 se diseñó para determinar si el sildenafil impactaba de manera favorable los síntomas de hipertensión pulmonar, al mejorar la función cardiopulmonar en pacientes con anemia de células falciformes. El objetivo primario a evaluar fue la prueba de caminata de 6min. Los participantes tenían anemia de células falciformes e hipertensión pulmonar de leve a severa, y fueron asignados al azar para recibir sildenafil o placebo durante 16semanas57,58.
Lamentablemente los resultados no fueron alentadores, pues hubo un aumento inexplicable de la estancia hospitalaria por crisis dolorosas en pacientes que recibieron sildenafil, hecho que motivó la finalización temprana del estudio (hospitalizaciones 45% sildenafil vs. 22% placebo, p=0,022)53. La terapia con inhibidores de la fosfodiesterasa5 solo se debe utilizar, de acuerdo con los resultados del estudio Walk-PHASST, en pacientes muy bien controlados con hidroxiurea y terapia de transfusión. Esta no se considera como terapia de primera línea58.
Disfunción ventricular izquierdaLas complicaciones cardiacas son comunes en pacientes con anemia de células falciformes. Los cambios estructurales en el área ventricular son consecuencia de mecanismos compensadores presentes como resultado de un estado de hipoxemia crónica, entre ellos, mayor volumen de plasma, incremento del gasto cardiaco con ligero aumento en la frecuencia cardiaca y cardiomegalia consecuente42.
El volumen de sangre que recibe el ventrículo izquierdo aumenta, lo que lleva a dilatación de esta cámara y somete a un gran estrés hemodinámico a la pared miocárdica, al tiempo que desarrolla hipertrofia excéntrica, con adelgazamiento y elongación de miofibrillas59.
La hipertrofia excéntrica inicialmente permite al ventrículo izquierdo adaptarse a una sobrecarga de volumen crónica porque la compliance diastólica está conservada, manteniendo presiones de llenado normales. A medida que la dilatación progresa, incrementa la masa miocárdica ventricular y se induce a hipertrofia muscular y aumento de grosor de la matriz colágena, provocándose alteración en las propiedades de relajación, distensibilidad y llenado del ventrículo izquierdo60.
Las condiciones de carga anormales llevan a dilatación de las cámaras y remodelación del miocardio con posterior progreso a disfunción ventricular. La contractilidad puede evaluarse a través de los índices de fase de eyección-fracción de acortamiento, fracción de eyección, velocidad de acortamiento circunferencial o intervalos de tiempo sistólicos.
La disfunción sistólica ventricular es rara. Los cribados ecocardiográficos en la mayoría de pacientes con anemia de células falciformes muestran función sistólica conservada61,62. La disfunción generalmente se observa en ancianos con comorbilidades asociadas, como hipertensión arterial e insuficiencia renal.
Nuevos estudios, como el de Denenberg et al.63 y el de Lamers et at.64, mostraron que sí hay disfunción contráctil miocárdica en pacientes con anemia de células falciformes. Hasta la fecha no existe consenso respecto a la proporción en la que se ve afectada la contractilidad por efectos inherentes de anemia severa de larga data o simplemente está alterada por sobrecarga de volumen crónico.
Ecocardiografía en anemia de células falciformesBraga et al.65 recientemente llevaron a cabo un estudio para determinar marcadores tempranos de disfunción cardíaca en pacientes con anemia de células falciformes a través de los índices de acortamiento (strain) y rotación (twist) ventricular, con el fin de determinar su relación con otros marcadores de riesgo cardiovascular.
La muestra de 40 pacientes con anemia de células falciformes y 40 controles sanos, a quienes se les realizó un análisis ecocardiográfico convencional, demostró aumentos significativos de volúmenes de masa ventricular y cámara cardiaca, así como en las presiones de llenado ventricular en el primer grupo de pacientes.
Contra todas las hipótesis y expectativas, al someter a ambos grupos a ecocardiografía bidimensional con técnica speckle-tracking, la fracción de eyección estaba conservada en ambos grupos, y adicionalmente no hubo diferencia entre los valores de deformidad (longitudinal, radial y circunferencial) del ventrículo izquierdo ni de deformidad longitudinal del ventrículo derecho entre estos. Sin embargo, la rotación del ventrículo izquierdo se vio disminuida en comparación con el grupo control. De igual forma, se establecieron correlaciones estadísticas entre la disminución en la rotación ventricular e índices de gravedad clínica altos, índice de volumen telediastólico del ventrículo izquierdo mayores, así como elevaciones de PsAP65.
Finalmente, puede concluirse que la rotación del ventrículo izquierdo podría considerarse como un parámetro de riesgo cardiovascular y herramienta útil en la detección temprana de los cambios incipientes en la función contráctil del miocardio en esta población.
Recientemente Barbosa et al.66 desarrollaron otro estudio con 90 pacientes con anemia de células falciformes, a quienes se les realizó ecocardiografía bidimensional con técnica speckle-tracking, y no hallaron diferencias en la función sistólica del ventrículo izquierdo o del derecho en comparación con los controles. A diferencia de otros estudios, en este no se aplicaron las medidas de rotación ventricular, lo cual podría haber marcado la diferencia de funcionabilidad miocárdica entre los grupos.
Monte et al.67 desarrollaron un estudio similar pero en pacientes con betatalasemia mayor, en donde tampoco hubo diferencias en los valores de deformación longitudinal, radial ni circunferencial en comparación con el grupo control, pero sí evidencia de disminución en los valores de torsión del ventrículo izquierdo en los pacientes con talasemia mayor.
De igual forma, Di María et al.68, llevaron a cabo un estudio entre niños tanzanos con anemia de células falciformes y controles de la misma edad, en el que participaron 213 pacientes vs. 49 controles, con el objetivo de evaluar si existen o no diferencias en la mecánica de rotación ventricular entre ambos grupos.
A través de ecocardiografía bidimensional speckle-tracking, en ambos grupos se evaluó la rotación basal y apical, el ángulo de torsión neta (diferencia instantánea en rotación entre el vértice y la base al final de la sístole), el pico de giro diferencial (diferencia máxima en rotación entre el vértice y la base independiente del tiempo) y la torsión y destorsión o untwisting (diferencia entre el retroceso apical y basal de retroceso).
Los resultados mostraron una disminución significativa en la rotación basal ventricular de los pacientes al compararlos con los controles. No se encontraron diferencias estadísticamente significativas en la rotación apical, torsión ni ángulo de torsión neta. La velocidad de rotación en vértice y base también fue más lenta en los pacientes con anemia de células falciformes. Otro de los datos de mayor relevancia es la tasa de destorsión pico media, que fue significativamente más lenta en los pacientes y tiene relación directa con la presencia de disfunción diastólica del ventrículo izquierdo68.
Para terminar, puede decirse que existe evidencia suficiente para avalar el desarrollo de cambios subclínicos en la función sistólica y diastólica del ventrículo izquierdo de niños con anemia de células falciformes, basándose en la mecánica de rotación ventricular, aunque su fracción de eyección o fracción de acortamiento esté preservada.
ConclusionesA pesar del aumento en la expectativa de vida de los pacientes con anemia de células falciformes en las últimas décadas, la longevidad ha puesto en evidencia un conjunto de anormalidades estructurales y funcionales en el plano cardiovascular y pulmonar principalmente, como consecuencia de la entidad subyacente, que conduce a una grave vasculopatía microvascular con una patogénesis que involucra disfuncionalidad endotelial, fenómenos inflamatorios, estado protrombótico y estrés oxidativo.
Múltiples estudios clínicos han incluido a estos pacientes con anemia de células falciformes, que a lo largo del tiempo desarrollan complicaciones como infarto agudo de miocardio, hipertensión arterial pulmonar, disfunción diastólica y/o sistólica ventricular, dentro de un grupo de alto riesgo donde la mortalidad es mucho mayor.
En consecuencia, la alta sospecha clínica de un infarto agudo de miocardio durante una crisis dolorosa marca la diferencia en el abordaje terapéutico, ya que las herramientas de estratificación de riesgo y modalidades diagnósticas son menos útiles que en el infarto agudo de miocardio típico.
La hipertensión arterial pulmonar, incluso con elevaciones moderadas, conlleva tasas de mortalidad elevadas; por tanto, la clave está en una selección de pacientes adecuada y pertinente, que conduzca a la realización de cateterismo cardiaco derecho, partiendo de estimaciones ecocardiográficas fiables de PsAP, basadas en la medición de VRT. Si bien la evidencia es escasa y se requieren más estudios para establecer recomendaciones, el manejo debe ir dirigido a la hidratación agresiva, a la oxigenación y al control del dolor y de la anemia. Para el control a largo plazo, la hidroxiurea sigue siendo la piedra angular para mejorar el curso de la enfermedad.
Se hace evidente también la superación de las limitaciones de la ecocardiografía convencional con las nuevas técnicas analíticas de acortamiento y rotación, que exponen cambios subclínicos en el miocardio de pacientes con falla cardíaca y fracción de eyección conservada.
Estas complicaciones cardiopulmonares contribuyen a una disminución considerable de la capacidad funcional, cada una de ellas de forma independiente, en donde la combinación de estas constituye un agravante clave del pronóstico.
Responsabilidades éticasDerecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Protección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.