


La miocarditis de células gigantes es una enfermedad rara, con gran impacto en cuanto a la mortalidad de pacientes usualmente jóvenes y sanos, que provoca diversas presentaciones clínicas como: la falla cardiaca, las arritmias ventriculares refractarias y los bloqueos. Su diagnóstico se establece con un alto índice de sospecha y con la confirmación por medio de la biopsia endomiocárdica por presencia de células gigantes multinucleadas sin presencia de un patrón granulomatoso. Su tratamiento inmunomodulador ha cambiado de manera importante en los últimos años, con una evidente disminución en los índices de mortalidad, aunque aún se requieren estrategias complementarias de manejo como el uso de dispositivos intracardiacos y/o el trasplante cardiaco para lograr mejores desenlaces a largo plazo.
Giant cell myocarditis is a rare condition with a great impact with regards to mortality of otherwise young and healthy patients, causing multiple clinical presentations, such as heart failure, refractory ventricular arrhythmias and blocks. Its diagnosis is reached with a high degree of suspicion and with the confirmation by means of a endomyocardial biopsy with the presence of multinucleated giant cells without a granulomatous pattern. Its immunomodulatory treatment has considerably changed during the last years, with a clear reduction in mortality rates, though complementary management strategies are still required, such as the use of intracardiac devices and/or heart transplantation in order to achieve better long-term results.
Un paciente de 27 años de edad previamente asintomático, quien ingresó a nuestra institución con diagnóstico de hospitalización reciente de taquicardia ventricular recurrente. El paciente refería un cuadro de 2 semanas de palpitaciones recurrentes, no irradiadas, asociadas a sensación de mareo por lo que consultó a otra institución donde le toman un EKG y evidencian taquicardia de complejo ancho que sugieren corresponde a taquicardia ventricular (TV) estable, iniciándose manejo con amiodarona sin lograr mejoría. Le realizaron ecocardiograma TT en el que se encontró deterioro de la FEVI hasta el 30%, con hipoquinesia difusa siendo llevado a angiografía coronaria descartándose lesiones significativas.
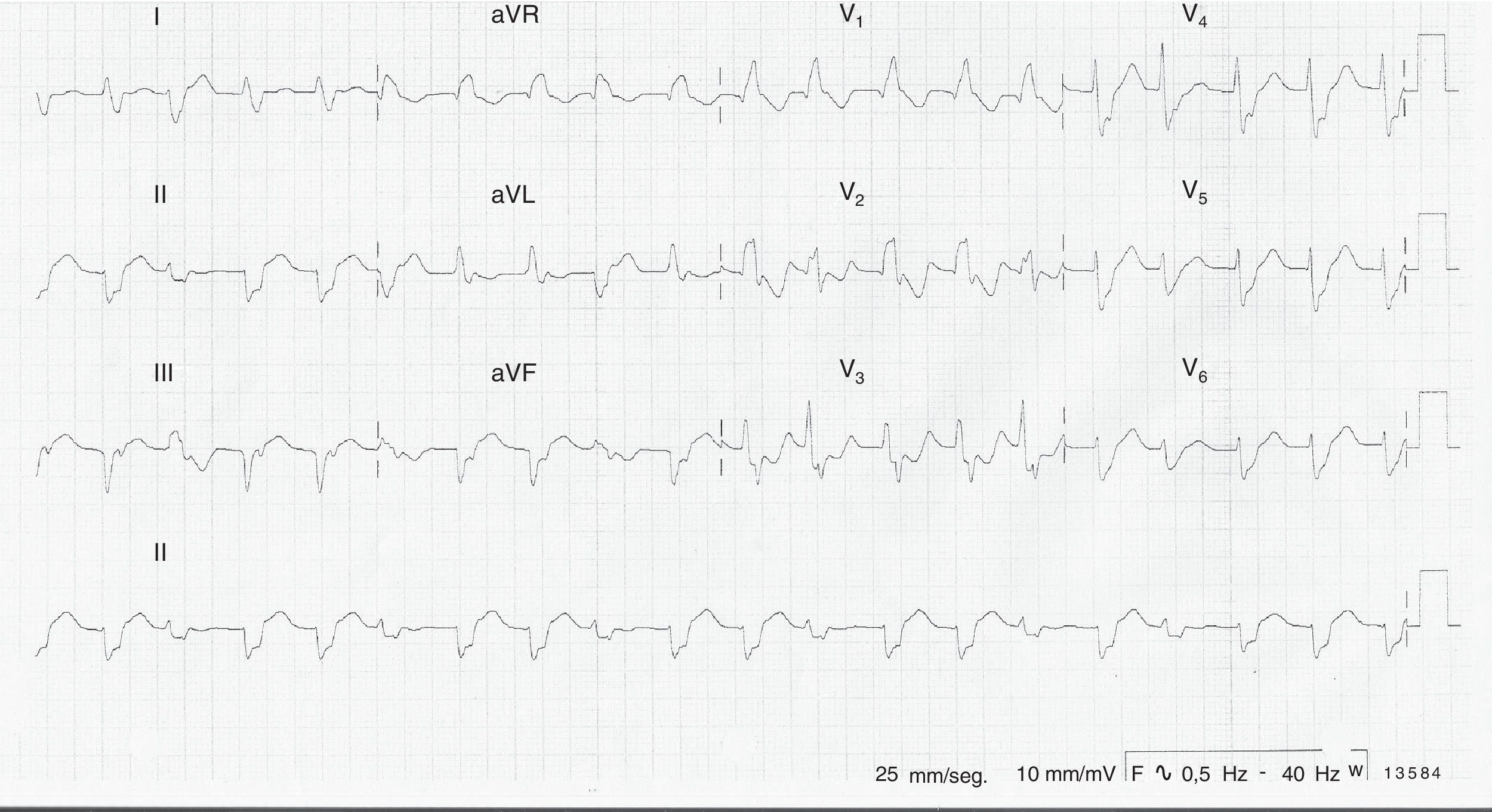
El paciente ingresó a nuestra institución por persistencia de síntomas, con hallazgos de taquicardia del complejo ancho y RR irregular que sugería TV polimórfica bidireccional (fig. 1). Se trasladó a la unidad de cuidado cardiovascular y se le realizaronn los exámenes paraclínicos con hallazgos de: troponina positiva (5.57) (VR 0.0–0.014), hemograma, azoados y electrolitos normales. Se descarta chagas y se solicitó perfil tiroídeo que salió normal.

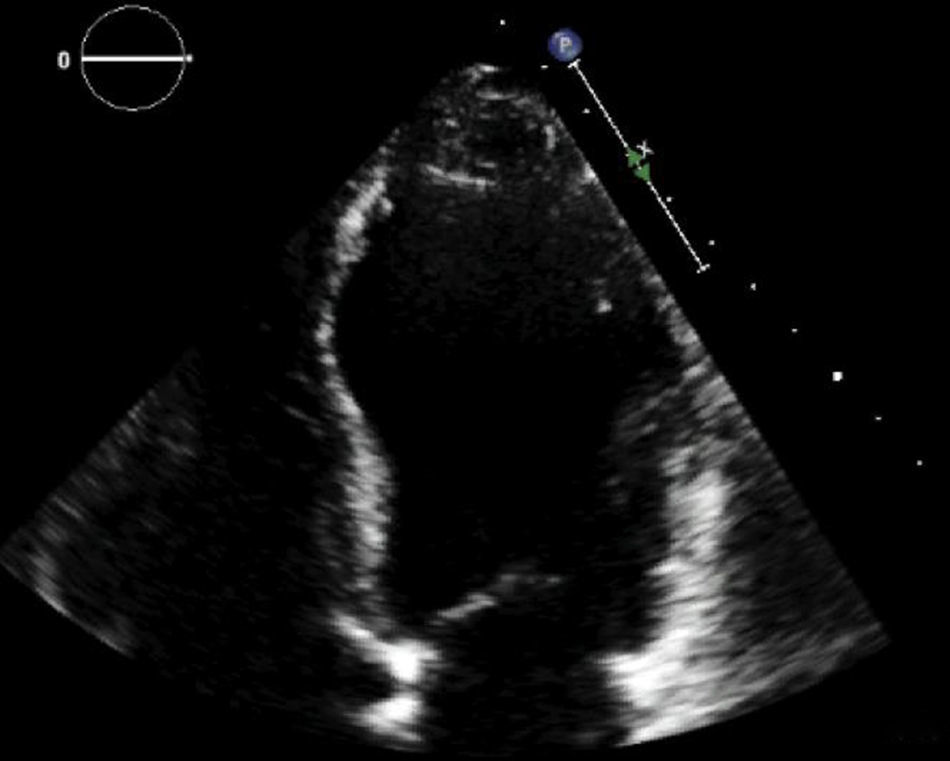
Se le realizó un ecocardiograma TT con hallazgos de: severa dilatación del ventrículo izquierdo (VFD: 100ml/m2), moderado compromiso de la función sistólica, FEVI 33% (fig. 2), aquinesia de los segmentos mediales del septum inferior, de la pared lateral y anterior, con hipoquinesia del resto de los segmentos. El strain longitudinal promedio fue -10%, observándose mayor compromiso de la mecánica ventricular de los segmentos mediales de todas las paredes. Se descarta compromiso valvular. El ventrículo derecho era normal, con función sistólica preservada.

Dada la presencia de trastornos segmentarios de la contractilidad y en el contexto de un paciente con arritmia ventricular y coronarias sanas, se decidió ampliar el estudio con resonancia cardiaca.
La resonancia cardiaca fue realizada en resonador Phillips Ingenia de 1,5T, obteniendo los siguientes resultados:
- -
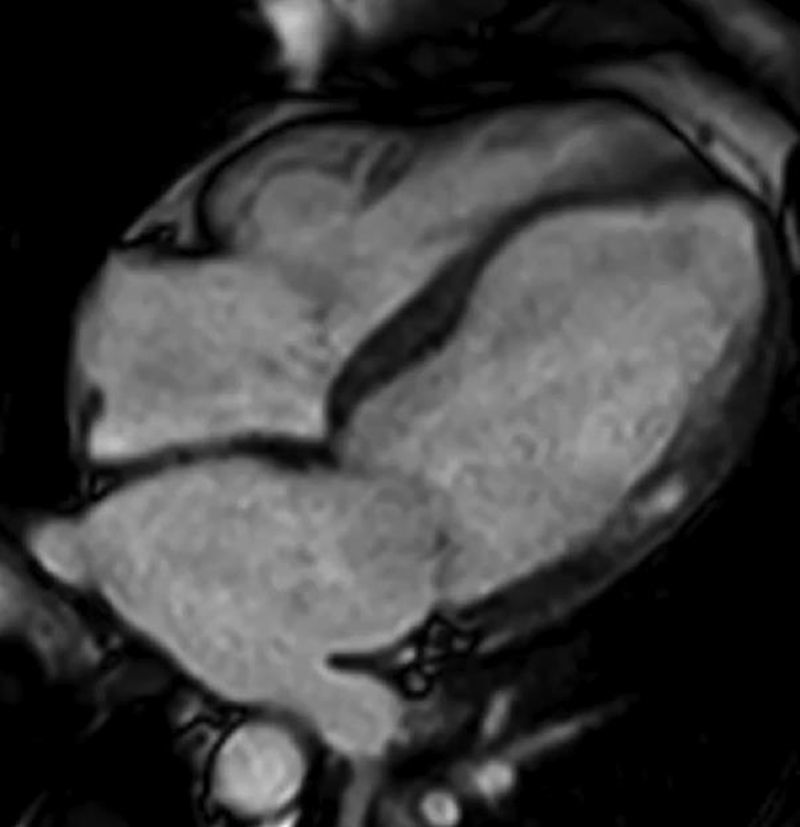
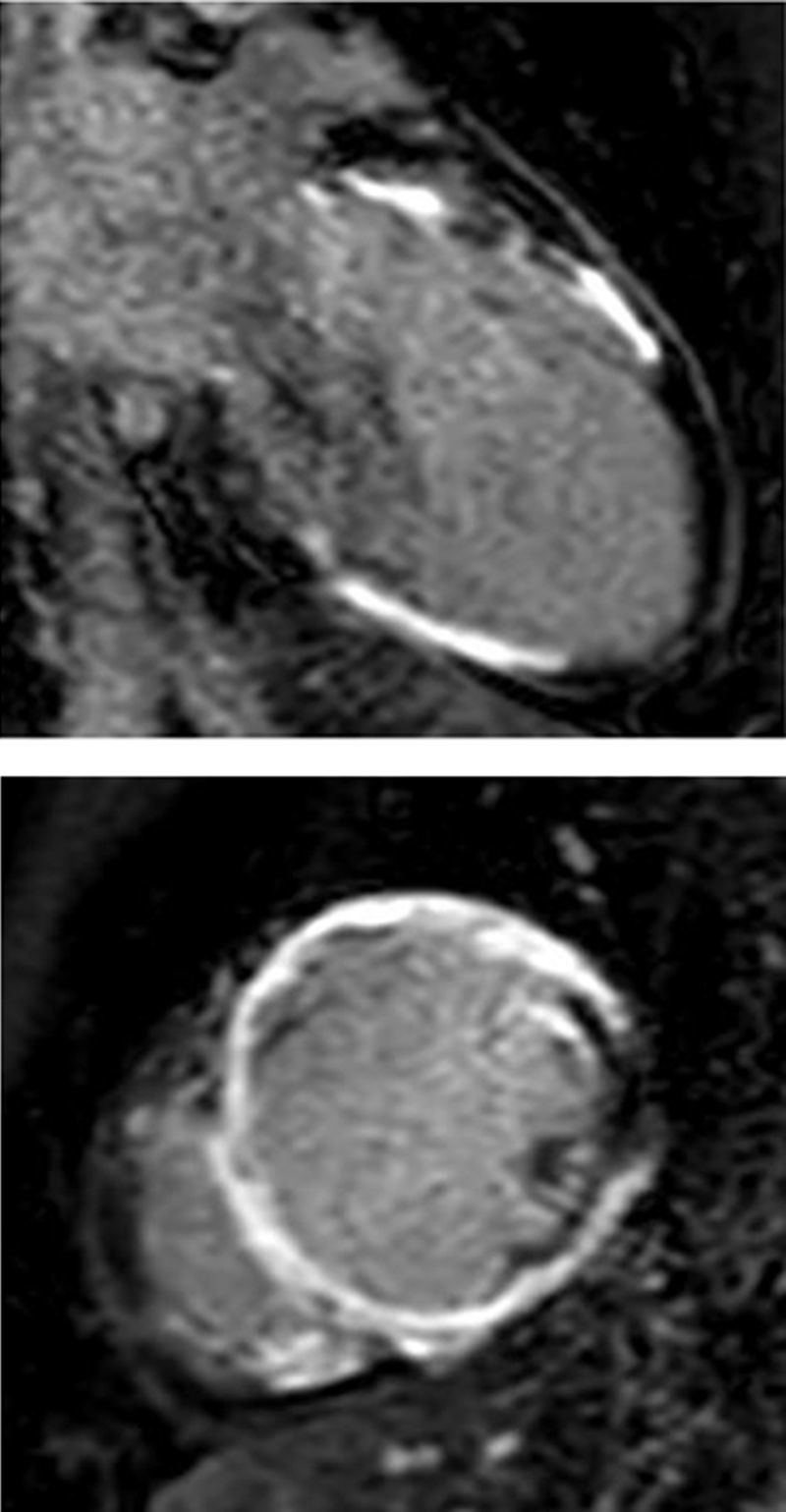
Ventrículo izquierdo severamente dilatado, severo compromiso de la función sistólica. Importante adelgazamiento y disquinesia de los segmentos mediales de todas las paredes y FE: 22% (fig. 3), edema miocárdico y depósito focal de gadolinio transmural en los segmentos mediales de todas las paredes y subepicárdico en los segmentos basales del septum (fig. 4).


Se consideró que estos hallazgos eran compatibles con la miocarditis, por lo que se decidió llevar al paciente a una biopsia endomiocárdica.
El paciente recae en taquicardia ventricular, presentando: episodios sostenidos, estables, polimórficos, asintomáticos; considerándose curso de TV incesante por lo que se inició manejo con amiodarona IV, definiéndose no candidato a modulación por extenso compromiso inflamatorio activo ni a uso de CDI por alto riesgo de descargas repetitivas o tormenta eléctrica.
El perfil viral fue negativo (citomegalovirus, rotavirus, adenovirus, influenza y parainfluenza), así como también el ELISA para VIH.
Fue llevado a biopsia endomiocárdica del ventrículo derecho, sin complicaciones, con reporte final de patología: “Endomiocardio con un área de extensa destrucción de miocitos y cambios reactivos. Estroma con abundante infiltrado de tipo linfoplasmocitario con eosinófilos, histiocitos y células gigantes multinucleadas. No se observaron inclusiones virales y las coloraciones ZN, Grocott, PAS y Giemsa fueron negativas para micobacterias, hongos y parásitos”.
Con estos hallazgos el diagnóstico definitivo fue: miocarditis de células gigantes.
El paciente fue evaluado en conjunto con el grupo de falla cardiaca decidiéndose iniciar con pulsos de: metilprednisolona, asociado a tacrolimus y micofenolato de mofetilo. El paciente siguió presentando episodios de taquicardia ventricular de diferentes morfologías y desarrollando el bloqueo completo de rama derecha. Durante su evolución presentó falla cardiaca con edema pulmonar, requiriendo ventilación no invasiva y levosimendan, con adecuada respuesta al manejo. Se le realizó un ecocardiograma de control sin cambios respecto al previo y resonancia cardiaca con ligera disminución del edema miocárdico, persistiendo el resto de hallazgos descritos.
Se presentó en junta médico-quirúrgica definiéndose la realización de trasplante cardiaco y plan de implante de dispositivo de asistencia ventricular en caso de nueva descompensación hemodinámica. El paciente entró en lista de trasplante, en emergencia 0, por inestabilidad eléctrica, presentando un nuevo episodio de taquicardia ventricular sostenida, que progresa a fibrilación ventricular. Se iniciaron maniobras de reanimación básica y avanzada con miras a iniciar asistencia ventricular, no obstante, a pesar de las maniobras el paciente falleció.
Revisión de la literaturaLa miocarditis se define como la infiltración del miocardio por tejido inflamatorio principalmente de los linfocitos. La miocarditis de células gigantes en una patología infrecuente, cuya historia natural, presentación, diagnóstico y pronóstico permanecen aún en evaluación. Los datos que se tienen hasta el momento provienen del reporte de pocos casos en la literatura.
El primer caso de la MCG se reportó en el año 1905 por Saltykov1, quien describió una miocarditis de curso fatal caracterizada por la presencia de células gigantes y un marcado infiltrado inflamatorio asociado a destrucción miocítica, la cual fue llamada miocarditis de células gigantes idiopática.
Es una enfermedad que se ha descrito clásicamente con tasas muy altas de mortalidad. En la serie más grande2, publicada hasta el momento, la mortalidad o necesidad de trasplante cardiaco a los 5,6 meses de seguimiento fue del 89%. El único factor modificador de la mortalidad demostrado fue la posibilidad de recibir manejo inmunosupresor, aumentando la sobrevida a 12,6 meses para aquellos pacientes que lo recibían, comparada con 3 meses para los que no recibían dicho tratamiento. Una serie más reciente de 32 casos de miocarditis de células gigantes confirmados por biopsia y/o estudio histológico del corazón explantado3, en el que el tratamiento inmunosupresor fue el tratamiento de elección, demostró que la mortalidad o la necesidad del trasplante disminuyó al 47% a un tiempo de seguimiento promedio de 11 meses desde el comienzo de los síntomas. Asimismo, las tasas de requerimiento del trasplante cardiaco calculadas fueron mucho menores que las anteriormente descritas, con tasas de sobrevida libre de trasplante del 69% a un año, el 58% a los dos años y el 52% a los 5 años. Considerándose, entonces que si bien la tasa de mortalidad y/o necesidad del trasplante cardiaco a corto plazo ha mejorado con el uso de la terapia médica, dichos resultados siguen siendo elevados en el contexto de una enfermedad altamente letal.
Su frecuencia de presentación es desconocida hasta el momento, sin embargo, se reconoce como una patología muy infrecuente (0.007% en Inglaterra hasta 0.051% en India), en la mayoría de los casos con diagnósticos definitivos provenientes de las autopsias o de los corazones explantados.
La miocarditis de células gigantes (MCG) es un fenómeno complejo, de origen multifactorial, en el que se han involucrado entre otros: las infecciones virales, las respuestas autoinmunes y/o la predisposición genética. Es así como reportes de casos han intentado enlazar la presencia de infecciones por diferentes agentes (herpes virus, Coxsakie virus b2, mycobacterium tuberculosis, parvovirus b19), como factor disparador del daño miocárdico4. También se ha descrito la participación de algunos mecanismos de autoinmunidad potenciados por la presencia de autoanticuerpos que tienen como blanco a la miosina cardiaca y que generan una reacción cruzada por mimetismo molecular, con sus receptores betaadrenérgicos. Esta hipótesis es la de mayor aceptación hasta el momento y ha ganado fortaleza debido a la respuesta obtenida en la disminución del daño miocárdico con el uso de la terapia inmunosupresora.
Una vez se activa el proceso de autoinmunidad se potencia un proceso de vasculitis mediado por quimioquinas, TLR (toll like receptors) y células dendríticas, moléculas que han sido encontradas en el corazón de pacientes con diagnóstico de MCG, ausentes en los controles sin la enfermedad.
La activación puntual de TLR5 favorece la inflamación perivascular y la producción de la citoquina CCL20. Por medio de TLR4 se medía la invasión por linfocitos T5,6. Por último, la influencia de factores genéticos de susceptibilidad ha sido investigada lográndose identificar hasta el momento 115 diferentes genes en corazones de pacientes con la MCG que parecerían estar implicados en la presentación de la entidad y al grado de daño miocárdico generado por la inflamación7. Otro hallazgo que refuerza la hipótesis de la presencia de una susceptibilidad genética, ha sido la identificación de alteraciones en la expresión de la plakoglobina (proteína estructural desmosomal) que enlaza a la MCG con otras entidades como la sarcoidosis cardiaca (SC) y la displasia arritmogénica del ventrículo derecho (DAVD), por lo que no se descarta que estas entidades compartan una susceptibilidad genética común.
Independientemente, del origen o el factor perpetuador de la enfermedad, el mecanismo final del daño miocárdico es secundario al compromiso inflamatorio de las células del músculo liso y el reclutamiento de fibras elásticas que llevan a la oclusión microvascular.
Por esta razón la MCG se considera una enfermedad inflamatoria mediada por linfocitos T y autoanticuerpos antimiosina. Se ha descrito una etapa temprana de infiltración de linfocitos T CD4 que producen una respuesta de tipo Th1, secretando IL2 e IFN gamma, y una posterior respuesta tardía Th2 que conlleva al desarrollo de fibrosis8.
Desde el punto de vista clínico es una enfermedad de pacientes jóvenes, previamente sanos, afectando de manera similar a hombres y mujeres, en la mayoría de los casos de raza blanca2.
Hasta en un 19% de los pacientes se ha descrito relacionada con patologías autoinmunes como: el LES, la artritis reumatoidea, la tiroiditis de Hashimoto, la miastenia gravis, la arteritis de Takayasu, el vitíligo, la anemia perniciosa, la colitis ulcerativa y la enfermedad de Crohn2,9, así como también se ha relacionado con la presencia de la hepatitis autoinmune y el lupus discoide.
Su presentación clínica más frecuente es la de falla cardiaca (aproximadamente en el 75% de los casos), pero también puede presentarse como taquicardia ventricular (15% de los casos), bloqueo AV (5%), simulando un evento coronario agudo (5% de los casos), síncope o muerte súbita2. Debe sospecharse en todo paciente que curse con una miocardiopatía dilatada de origen no determinado, de reciente aparición, en quienes se hayan descartado las causas más frecuentes.
La falla cardiaca suele presentarse como una entidad de difícil manejo, rápidamente progresiva y refractaria a la terapia médica convencional, lo que lleva a una alta tasa de requerimiento de soportes invasivos en este grupo de pacientes y que se relaciona de manera directa con el pronóstico sombrío. De un grupo de 26 pacientes con diagnóstico confirmado de la MCG, 7 de los pacientes del grupo de terapia médica cursaron con FEVI entre el 20 y el 40%, con CF NYHA entre II y IV, 6 de los cuales requirieron trasplante cardiaco3.
La presentación como TV sostenida o no sostenida es la segunda en frecuencia y también se relaciona con una mayor probabilidad de muerte o requerimiento de trasplante cardiaco. Se presenta también, en alto porcentaje, en el grupo de pacientes que debutan con falla cardiaca. En una serie de 32 pacientes, se documentó la presencia de uno o más episodios de TV, en el 92% de los pacientes, siendo sostenida en el 65%. A mediano y largo plazo 18 pacientes de este grupo requirieron implante de CDI3.
Dada la aparente mejoría presentada en la mortalidad en los pacientes con MCG a corto plazo, la tasa de implante de CDI es una de las características que ha cambiado con el tiempo ya que en el registro del año 1997 solo 12% de los pacientes recibieron un CDI comparado con el 56% del total y el 69% de los pacientes en grupo médico que lo recibieron en este último estudio. Sin descripción de tormentas arrítmicas en este grupo de pacientes quienes recibieron dispositivos3.
La presentación clínica como bloqueo AV se suele relacionar más con la sarcoidosis cardiaca, también se ha documentado en pacientes con la MCG. En un estudio reciente, 25% de los pacientes con este diagnóstico requirieron el implante de un marcapasos10. Y así mismo de todos los pacientes jóvenes (menores de 55 años de edad) que se presentaron con bloqueo AV idiopático como síntoma predominante de su enfermedad, presentaron por lo menos en un 25% diagnóstico de la SC o la MCG en la biopsia endomiocárdica.
El hemograma, los perfiles virales o pruebas de detección serológica carecen de sensibilidad en el diagnóstico y solo deberán realizarse con el objetivo de descartar otras entidades de comportamiento clínico similar. Las ayudas imagenológicas han logrado un importante desarrollo en este campo, favoreciendo el diagnóstico más temprano de la entidad.
Los hallazgos ecocardiográficos en pacientes con la MCG pueden ser variables e incluyen desde la presencia de trastornos segmentarios de la contractilidad que no corresponden a un territorio coronario anatómico, en los ventrículos de tamaño normal, hasta el adelgazamiento de la pared miocárdica, con deterioro progresivo de la función sistólica. La función ventricular puede verse deteriorada aún en cuestión de pocos días. El compromiso del ventrículo derecho usualmente sigue al compromiso del ventrículo izquierdo11.
Dada la excelente resolución de la resonancia cardiaca, esta técnica permite objetivar aún mejor la función sistólica de ambos ventrículos y la identificación de los segmentos comprometidos. La posibilidad de caracterizar el tejido mediante secuencias específicas, permite evidenciar los procesos inflamatorios, que aunque inespecíficos, orienten el diagnóstico. Así mismo, las secuencias de realce tardío mediante la utilización del gadolinio permiten identificar las zonas de aumento en el espacio extracelular, que en este caso representa el infiltrado inflamatorio y la destrucción de los miocitos. No hay un tipo de realce específico para esta patología, no obstante, su hallazgo permite identificar en la mayoría de los casos, el severo compromiso miocárdico y de esta manera servir como guía para la realización de la biopsia miocárdica, buscando disminuir los errores de muestreo. Las nuevas secuencias de T1 mapping que aún se encuentran en investigación, pero que ya han demostrado su utilidad en algunas patologías, permitirán objetivar aún más el compromiso miocárdico e incluso calcular el volumen extracelular, que tendría que estar aumentado en estos pacientes.
Las imágenes de medicina nuclear como SPECT o PET, así como la TAC son usadas en el contexto de otras entidades granulomatosas como la sarcoidosis cardiaca y no han tenido el impacto demostrado en la aproximación diagnóstica o de seguimiento de los pacientes con la MCG11.
La introducción de la biopsia endomiocardica (EBM) hacia finales del siglo veinte logró un cambio muy importante para la ampliación del conocimiento en la miocardiopatía dilatada y más aún para la identificación de la MCG. El desarrollo de métodos de identificación celular por marcadores directos o inmunohistoquímica ha permitido aumentar la sensibilidad de la biopsia para el diagnóstico de la miocarditis.
Los hallazgos histológicos consisten en áreas serpiginosas de necrosis miocárdica mezcladas con infiltrado inflamatorio mixto compuesto de: los linfocitos, las células plasmáticas, los histiocitos, los eosinófilos y las células gigantes multinucleadas. No existe formación definida de los granulomas y las células gigantes, usualmente tienen un origen histiocítico. Adicionalmente, se observa necrosis y destrucción miocítica, como hallazgo importante en esta patología. La identificación por inmunohistoquímica de linfocitos T ha permitido evidenciar el predominio de poblaciones clonales de linfocitos T CD 8+ Al realizar una comparación con la SC desde el punto de vista de las características histológicas, se encuentra que en esta última existe mayor formación de granulomas, menor tasa de presencia de los eosinófilos, ausencia de la necrosis miocítica y la población de linfocitos de predominio CD4+12.
Algunas características de la biopsia endomiocárdica de pacientes con la MCG son compartidas parcialmente con otras entidades sistémicas (granulomatosis de Wegener, miocarditis de hipersensibilidad, TBC y miocarditis reumática)12, por lo que se requiere un conocimiento amplio por parte del patólogo, ya que cada entidad tiene un pronóstico y enfoque terapéutico completamente distinto.
La sensibilidad de las biopsias varía en las series analizadas. En los estudios más antiguos, la sensibilidad es del 65% aproximadamente, logrando aumentarse al 85-90%, en estudios más recientes.
La tasa de diagnóstico logrado por la biopsia en el estudio del Dr. Kandolin3, es de lejos mucho mejor que la reportada en el estudio del Dr. Cooper2, lográndose establecer un diagnóstico en el 82% (26/32) de los pacientes vs. el 56% en el del Dr. Cooper. El 27% de los diagnósticos fueron hechos con dos o más muestras.
Los riesgos de la biopsia son bajos, con una posibilidad de muerte de 1:1000 y de perforación ventricular de 1:25013.
Es claro que la incorporación de estudios de RMN y PET para el diagnóstico, y como guía para la realización de las biopsias endomiocárdicas, han impactado de manera muy positiva en el aumento de la sensibilidad de las biopsias3,14; incluso identificando los pacientes que requieren la realización de biopsias por toracotomía.
Las biopsias del ventrículo izquierdo (VI) generan información diagnóstica crucial en caso de compromiso aislado de este. Existe una publicación del año 201315, donde se analizan un total de 4.221 pacientes llevados a biopsia (1.153 con BEM del VI, 672 con BEM del VD y 2.369 con biopsia biventricular), encontrando más baja la tasa de complicaciones para la toma de biopsia del VI en comparación con el derecho (0.33% vs. 0.45%) probablemente explicado por el grosor de la pared miocárdica15. Asimismo, con un mayor rendimiento en cuanto el establecimiento del diagnóstico en desórdenes que afectaban predominantemente el VI (97.8% vs. 53%)16.
El diagnóstico diferencial más frecuente de la MCG es el de sarcoidosis cardiaca ya que en estas dos entidades se presenta compromiso granulomatoso, no obstante, como se mencionó previamente el infiltrado inflamatorio de la sarcoidosis es mucho más organizado, con formación de granulomas, mayor fibrosis y menor daño tisular. Desde el punto de vista clínico también existen importantes diferencias entre estas dos entidades que nos permiten una mayor orientación. La preferencia en la SC es hacia la raza negra con tasas que han sido descritas hasta de un 31%, comparada con solo un 4% de pacientes de raza negra con la MCG. La presentación clínica como síncope y/o bloqueo AV es más frecuente en la SC que en la MCG (31% vs. 5% y 50% vs. 15%, respectivamente)13. Los episodios de TV o FV se han reportado con frecuencias similares en las dos entidades. La duración de los síntomas antes de la presentación fue mayor para la SC que para la MCG (5.5 meses vs. 1.2 meses) sin diferencias demostradas en la edad de presentación o género13.
El manejo médico de la enfermedad incluye el manejo convencional para falla cardiaca. En la MCG aguda el deterioro clínico puede presentarse en cuestión de horas con requerimiento de los marcapasos temporales o la terapia inotrópica o en el peor de los casos, los dispositivos de asistencia ventricular13.
La terapia inmunosupresora disminuye de manera marcada la mortalidad y aumenta el tiempo de sobrevida libre de trasplante3. Un registro multicéntrico2, reportó 63 pacientes con diagnóstico confirmado de la MCG. Los pacientes que no recibieron inmunosupresión tuvieron una sobrevida promedio de 3 meses. En este estudio varios pacientes (33/63) recibieron algún tipo de terapia: 11 pacientes recibieron monoterapia con corticoesteroides; 11 pacientes azatioprina con esteroides; 3 pacientes, ciclosporina combinada con corticoides; 5 pacientes terapia combinada con azatioprina; y 3 con corticoides, azatioprina y muromonab, logrando un mejoría en la sobrevida a 12.3 meses.
Posterior al estudio de seguimiento de Cooper, se identificó que el someter a estos pacientes a la inmunosupresión podría mejorar los desenlaces a largo plazo, sin embargo, este únicamente pudo ser evaluado en un estudio prospectivo hasta el año 2008, donde 11 pacientes fueron sometidos a terapia con inmunosupresión basada en: ciclosporina, asociada a terapia con esteroides y/o 10 días de muromonab–CD3 en casos de pacientes con la MCG probados por biopsia y con menos de tres meses de inicio de síntomas, 4 de los cuales fueron aleatorizados a los dos brazos de terapia y 7 hicieron parte de un registro de tratamiento. Se realizó un seguimiento con cambios en la FEV y con biopsias rutinarias al ingreso y a las 4 semanas de posterapia, en las que se observó la actividad de la enfermedad. Seis pacientes recibieron manejo con CDI en su seguimiento por presencia de TV, 1 paciente falleció por la enfermedad, dos requirieron trasplante a 1 año de observación y de los 10 sobrevivientes no se evidenció cambio en la FEVI al mes y un paciente que suspendió la inmunosupresión al año de seguimiento, falleció por recurrencia de la enfermedad probada por la biopsia17.
Basados en los datos disponibles a la actualidad se considera que la terapia con ciclosporina es la terapia de elección, considerándose el papel protagónico de los linfocitos T en esta entidad, no obstante, un estudio describe la necesidad de uso de terapia inmunosupresora no convencional en un paciente quien presentó recaída posterior a la terapia inicial por lo que se definió el manejo con una combinación de: tacrolimus, mofetil micofenolato y prednisona por ser esta una terapia usada para la inmunosupresión en el paciente postrasplante demostrándose que cerca de 1 año posterior al uso de esta terapia el paciente se encontró estable e incluso con discreta mejoría de la FEVI, por lo que se propone este esquema como una terapia alternativa al uso convencional de la ciclosporina en combinación, como terapia de base para esta entidad18.
Así mismo, la terapia de inmunosupresión alternativa con la globulina antitimocítica de conejo a dosis de 1.5mg/k se probó en combinación con los esteroides en un pulso de 7 días, posteriormente, asociada al uso de tacrolimus y MMF disminuyendo la actividad inflamatoria de la enfermedad, en un paciente que aproximadamente 6 meses después al inicio de la terapia requirió ser llevado a trasplante cardiaco19.
Se conoce que la miocarditis de células gigantes (MCG) es una entidad que recurre en el corazón trasplantado aunque su tasa de recurrencia es desconocida, con un tiempo promedio de aparición en el corazón trasplantado de 3.0 años2.
En el estudio de Cooper se describen 34 pacientes quienes fueron llevados a trasplante cardiaco, en los que se presentó recaída en 9 pacientes de los 34 en el seguimiento por la biopsia. Tres de estos pacientes recurrieron con síntomas de falla cardiaca, de los cuales 1 murió y los otros dos respondieron a la modificación de la terapia inmunosupresora. Los otros 6 presentaron recurrencia asintomática de la enfermedad en las biopsias de seguimiento a un promedio de 2.1 años posterior al trasplante20. Así, entonces la posibilidad de recurrencia no debe ser una contraindicación para el manejo con trasplante.
La presentación de recurrencia asintomática, de hecho no confiere un pronóstico grave. De 5 pacientes seguidos en un estudio, 3 presentaron la MCG recurrente asintomática a los 13 meses posteriores al trasplante, en quienes la optimización de la terapia inmunosupresora logro un adecuado control lo cual fue evidenciado en las biopsias de seguimiento que evidenciaron tal resolución20.
El uso de soporte ventricular como puente a trasplante en estos pacientes ha sido descrito por diferentes grupos y este se ha enlazado como un factor predictor independiente de probabilidad de recaída en el injerto8.
En un estudio dirigido a evaluar los desenlaces de uso de dispositivos de asistencia ventricular como terapia puente a trasplante, realizado de manera retrospectiva de pacientes con confirmación histológica de la MCG, se encontraron 6 pacientes que recibieron asistencia ventricular, con edad promedio de 44 ± 18 años. Cinco de los 6 pacientes fueron llevados a soporte biventricular y uno solo fue llevado a asistencia ventricular izquierda. De estos dos pacientes murieron durante el uso de la asistencia. Los cuatro adicionales fueron llevados a trasplante ortotópico en un promedio de 77 ± 42 días después del inicio del soporte. Los 4 pacientes que lograron ser llevados a trasplante cardiaco continuaron vivos hasta el final del seguimiento a 5.7 ± 4.1 años. Dos pacientes tuvieron recurrencia de la MCG en el injerto, los cuales requirieron ajuste de la terapia de inmunosupresión con adecuada respuesta. Tres pacientes tuvieron alto grado de rechazo a los 66 ± 52 días del injerto. La función ventricular se conservó en todos los pacientes y solo un paciente desarrolló vasculopatía del injerto21.
Poco es conocido en relación a la evolución de los pacientes a largo plazo. Un reciente estudio publicado en American Journal of Cardiology tomó a los pacientes con diagnóstico confirmado de la MCG que hubiesen sobrevivido más de 1 año en manejo médico, sin trasplante cardiaco para evaluar los desenlaces, encontrando que 15/26 pacientes recibieron manejo con CDI, existiendo descenso posterior al año de la terapia inmunosupresora de dos medicamentos a un medicamento inmunosupresor en promedio. Se observó el curso de fenómenos infecciosos en 13 pacientes (41 eventos). Tres pacientes murieron, 5 fueron llevados a trasplante y 23/26 continuaron vivos al seguimiento (5.5 años). La sobrevida libre de muerte, el trasplante, las arritmias ventriculares y la falla cardiaca fue de 35% a los 5 años. No existieron nuevos pacientes con arritmias ventriculares, aquellos que se presentaron clínicamente con arritmias el primer año fueron quienes continuaron con la presentación de estas22.
El papel del soporte circulatorio o asistencia ventricular como soporte a recuperación o trasplante cardiaco, la óptima duración de la inmunosupresión en pacientes quienes recuperan la función ventricular y la mejor estrategia de manejo para la prevención de recurrencia de la enfermedad en el corazón trasplantado siguen siendo motivo de debate y hasta el momento no hay directrices generadas sobre tales asuntos, siendo estos temas interesantes oportunidades para el desarrollo de investigaciones futuras17.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesDeclaramos expresamente no tener ningún conflicto de intereses en la elaboración del presente trabajo.