



La hipertensión pulmonar es un desorden complejo que requiere manejo multidisciplinario. Recientes avances médicos han llevado al reconocimiento de nuevas terapias que ofrecen alternativas de tratamiento, como se concluye a partir de estudios clínicos publicados en el último año.
Esta revisión del tema discute los ensayos clínicos que han dado lugar a la aprobación de nuevos fármacos para el tratamiento de la hipertensión pulmonar. Dos estudios clínicos fase tres, controlados, aleatorizados demostraron que el riociguat, un estimulador de la guanilato ciclasa soluble, mejoró significativamente la capacidad de ejercicio, la resistencia vascular pulmonar, el nivel de NT-proBNP y la clase funcional tanto en pacientes con hipertensión pulmonar tromboembólica sin indicación de manejo quirúrgico, como en pacientes con hipertensión arterial pulmonar sintomática sin tratamiento o que estaban recibiendo antagonistas del receptor de la endotelina o prostanoides.
Así mismo, el macitentán, un antagonista dual del receptor de endotelina redujo la morbimortalidad en forma dosis-dependiente en pacientes con hipertensión arterial pulmonar en un periodo de 3,5 años. Los resultados de estas investigaciones adicionan alternativas a la aproximación terapéutica de la hipertensión arterial pulmonar como se observa en las nuevas guías de hipertensión pulmonar realizadas en Niza, Francia, publicadas en 2013. Aún es indispensable conducir nuevos ensayos clínicos que comparen estas moléculas con el tratamiento recomendado hoy en día.
Pulmonary hypertension is a complex disorder that requires a multidisciplinary approach. Recent medical advances have led to the recognition of new therapies that offer management alternatives as concluded from clinical studies published in the past year.
This topic review discusses the clinical trials that led to approval of new drugs for the management of pulmonary hypertension. Two phase three trials showed that riociguat, a stimulator of soluble guanylate cyclase, significantly improved exercise capacity, pulmonary vascular resistance, NT-proBNP levels and functional class both in patients with thromboembolic pulmonary hypertension without indication of surgical treatment and in symptomatic pulmonary arterial hypertension patients who were receiving endothelin receptor antagonists or prostanoids.
Macitentan, a dual endothelin receptor antagonist reduced morbidity and mortality in a dose dependent manner in patients with hypertension in a period of 3.5 years. The results of these investigations offer an alternative therapeutic approach to pulmonary arterial hypertension as outlined in the new guidelines for pulmonary hypertension performed in Nice, France published in 2013. It is still necessary to conduct new clinical trials comparing these new molecules with the treatment that is currently recommended.
La hipertensión pulmonar es un trastorno complejo que puede ser idiopático o estar relacionado con distintas entidades. Es una enfermedad de las arterias pulmonares de pequeño calibre que se caracteriza por la proliferación y remodelación vascular, fenómeno que se traduce en un aumento progresivo de la resistencia vascular pulmonar y, en última instancia, en insuficiencia ventricular derecha y muerte1,2.
La investigación sobre hipertensión pulmonar ha avanzado enormemente en la última década, de tal modo que se han desarrollado distintas alternativas de tratamiento para mejorar la supervivencia y calidad de vida de los pacientes que la padecen.
Pese a la disponibilidad de múltiples medicamentos aprobados en la actualidad, no existían fármacos aprobados para la hipertensión pulmonar tromboembólica crónica (HTPTEC), ni seguimiento a largo plazo para el manejo de la hipertensión pulmonar arterial sintomática, hecho que motivó el desarrollo de investigaciones acerca de nuevos medicamentos dirigidos al tratamiento de la hipertensión pulmonar.
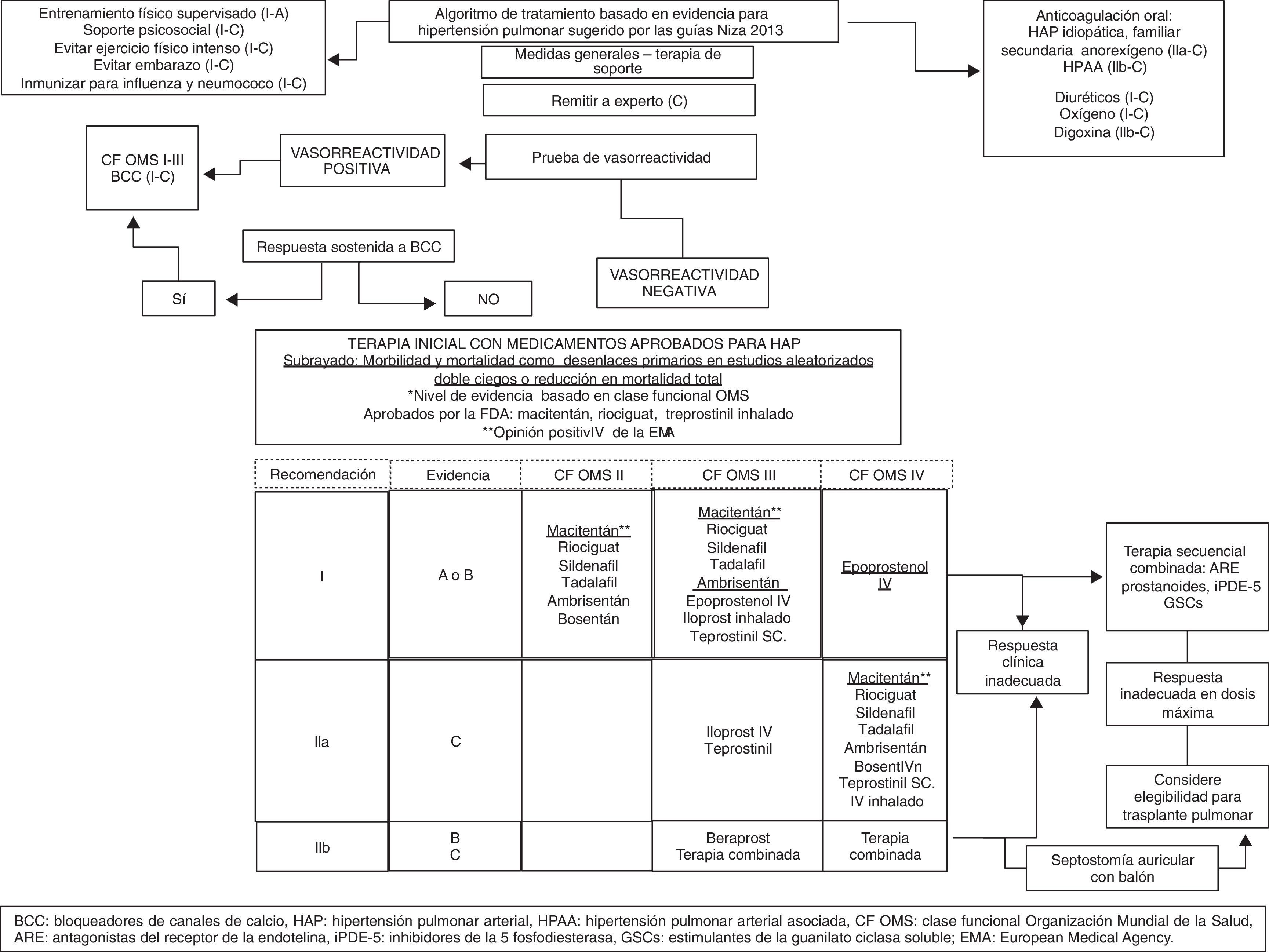
DiagnósticoLos principios fundamentales para el diagnóstico de hipertensión pulmonar están ampliamente descritos y fueron ratificados en el último simposio mundial de hipertensión pulmonar. Esta se debe sospechar en cualquier paciente con disnea sin causa establecida, síncope y/o signos de falla ventricular derecha. Los autores de las guías recientes han modificado el algoritmo diagnóstico (fig. 1), al agregar la capacidad de difusión del monóxido de carbono en la evaluación inicial, puesto que la espirometría no siempre demuestra la presencia de enfermedad del parénquima pulmonar en pacientes con fibrosis pulmonar y/o enfisema.

Algoritmo de tratamiento para el manejo de la hipertensión pulmonar.
Adaptado y modificado de: Galie et al.2
La hipertensión pulmonar se refiere a la presencia de presión anormalmente alta en la circulación pulmonar. El diagnóstico confirmatorio requiere la realización de un cateterismo derecho. La hipertensión arterial pulmonar es un diagnóstico de exclusión. La actual definición hemodinámica consiste en una presión media de la arteria pulmonar (PAPm) mayor o igual a 25mm Hg en reposo; presión en cuña capilar pulmonar –presión de la aurícula izquierda o presión de fin de diástole– ≤ 15mm Hg. Con el fin de simplificar la definición de hipertensión arterial pulmonar, las guías ya no recomiendan incluir la resistencia vascular pulmonar debido a que, en ciertas poblaciones con hipertensión pulmonar, esta se mantiene dentro de los valores normales. Por tanto, se debe incluir la resistencia vascular pulmonar en la definición hemodinámica, únicamente en los pacientes con hipertensión arterial pulmonar precapilar (PAPm > 25mm Hg, PCP ≤ 15mm Hg, y resistencia vascular pulmonar > 3 WU.
Algunos aspectos acerca de la definición de hipertensión pulmonar, tratados en el quinto simposio mundial de hipertensión pulmonar, llevado a cabo en Niza en 2013, ameritan mayor investigación. Por ejemplo, no es claro si se debe introducir el término «hipertensión pulmonar limítrofe» en pacientes con PAPm entre 21 y 24mm Hg, puesto que se desconocen las implicaciones, los pronósticos y las terapéuticas. Sin embargo, los autores reconocen que se debe hacer un seguimiento cuidadoso, en especial en población de alto riesgo para desarrollar hipertensión pulmonar. Otro tema pendiente por aclarar es la diferenciación entre hipertensión arterial pulmonar e hipertensión pulmonar debido a falla cardiaca izquierda con fracción de eyección preservada. En este sentido, la hipertensión pulmonar inducida por el ejercicio y la prueba de carga con fluido, pueden identificar a los pacientes con insuficiencia cardiaca y fracción de eyección preservada pero con presión en cuña normal y así reducir los diagnósticos inadecuados de hipertensión arterial pulmonar. Aún está pendiente estandarizar estas pruebas para recomendarlas en la práctica clínica1.
La disnea es el síntoma más común en estos pacientes; otras manifestaciones inespecíficas incluyen fatiga, presíncope y síncope. En el examen físico es frecuente encontrar acentuación del S2 e insuficiencia tricúspide durante la auscultación cardiaca. También puede presentarse ascitis, ingurgitación yugular y edema de miembros inferiores3.
Pese a que cada vez hay mayor conciencia de la importancia de diagnosticar la hipertensión pulmonar, a menudo hay una brecha entre la aparición de los síntomas y el diagnóstico. El registro REVEAL4 mostró que un 21% de los pacientes con hipertensión pulmonar idiopática presentaron síntomas dos años previos al diagnóstico. La mayoría de estos pacientes tenía clase funcional iii o iv, lo cual predice un pobre desenlace. Por esto, los expertos insisten en que el diagnóstico temprano es benéfico ya que ofrece terapias que retardan la aparición de la falla cardiaca derecha.
Los pacientes que padecen enfermedades del tejido conectivo, enfermedades congénitas cardiovasculares, hepatopatía crónica, infección por el virus de inmunodeficiencia humana o hipertensión arterial pulmonar familiar se benefician de un diagnóstico temprano. El progreso más importante se ha llevado a cabo en el tamizaje de los pacientes con esclerodermia sin síntomas sugestivos de hipertensión pulmonar, el cual incluye un examen físico meticuloso en búsqueda de telangiectasias, anticuerpos anticentrómero, pruebas funcionales pulmonares, capacidad de difusión de CO, ecocardiograma y marcadores (NT-proBNP y ácido úrico en la etapa inicial)1.
El estudio inicial más apropiado para evaluar pacientes con posible hipertensión arterial pulmonar es el ecocardiograma transtorácico, que ofrece la posibilidad de calcular la presión sistólica de la arteria pulmonar (PSAP) y derivar de esta la PAPm (PAPm=0,65 presión sistólica de la arteria pulmonar+0,55mm Hg). También permite hacer seguimiento de la respuesta al manejo terapéutico e identificar factores pronósticos5.
EpidemiologíaAntes se consideraba a la hipertensión arterial pulmonar como una entidad rara. Un registro nacional francés sugiere una prevalencia de aproximadamente quince casos por millón. La hipertensión arterial pulmonar idiopática es la forma más común, más frecuente en mujeres (tasa mujer: hombre de 1,7:1) y edad promedio de 37 años en el momento del diagnóstico. Estudios recientes muestran que el rango de edad de los individuos afectados ha aumentado; se han reportado múltiples casos en mayores de 70 años. Una posible explicación del cambio en el perfil clínico de la hipertensión arterial pulmonar es el incremento en el conocimiento y la disponibilidad de nuevas terapias4.
El 20% de los pacientes con hipertensión pulmonar idiopática y el 70% de los pacientes con hipertensión arterial pulmonar familiar tienen una mutación en el gen del receptor II de la proteína morfogenética ósea (BMPR2). Estos hallazgos aumentan la posibilidad de crear un tamizaje familiar. Sin embargo, esta entidad tiene baja penetrancia, pues el riesgo de desarrollar hipertensión arterial pulmonar es cercano al 20% en los pacientes con la mutación del BMPR26.
PronósticoEl pronóstico de estos pacientes depende de la etiología de la hipertensión arterial pulmonar. Aunque ha mejorado sustancialmente en la última década, el pronóstico general es pobre. La mortalidad al año es aproximadamente del 15% en los pacientes que reciben terapia. En la cohorte de hipertensión arterial pulmonar idiopática del registro francés, la supervivencia al año fue del 88%, en comparación con la sobrevida de hipertensión arterial pulmonar secundaria a enfermedad autoinmune y VIH, que fue del 67 y del 58%, respectivamente3.
La prueba vasodilatadora aguda tiene un valor pronóstico importante y se debe hacer en los pacientes con hipertensión arterial pulmonar idiopática, posibles candidatos a recibir tratamiento con bloqueadores de canales de calcio a largo plazo7.
Los factores que predicen un pobre desenlace incluyen: disminución de la clase funcional de la Organización Mundial de la Salud (OMS), pobre tolerancia al ejercicio determinada por la caminata de 6 minutos, aumento de la presión de la aurícula derecha, disfunción ventricular derecha significativa, índice cardiaco bajo, elevación del péptido natriurético cerebral (BNP) y esclerodermia8.
Dado que la función ventricular derecha es el principal factor para determinar supervivencia, se ha propuesto que la resonancia magnética cardiaca (hoy en día el estándar de oro para investigar la estructura y los volúmenes cardiacos) tendría un valor pronóstico importante en hipertensión arterial pulmonar9.
FisiopatologíaExiste un amplio rango de condiciones médicas y ambientales que se asocian con el desarrollo de hipertensión arterial pulmonar, de ahí que sean múltiples los mecanismos fisiopatológicos. Algunas teorías se basan en modelos moleculares y genéticos que estudian la cascada de inflamación, el músculo liso, las células endoteliales y la adventicia. La causa principal del aumento de la resistencia vascular pulmonar es la pérdida de luz vascular debida a la remodelación vascular producida por un desbalance entre la proliferación y la apoptosis. El desequilibrio entre vasoconstricción y vasodilatación también cumple un papel fundamental y, por ende, es la base del tratamiento farmacológico actual10.
Se han estudiado distintas moléculas relacionadas con la aparición de hipertensión arterial pulmonar. Las prostaciclinas y el tromboxano A2 son metabolitos del ácido araquidónico, ampliamente estudiadas en la patogénesis de esta entidad. Por un lado, la prostaciclina PGI2 es un potente vasodilatador: inhibe la agregación plaquetaria y tiene propiedades antiproliferativas. En contraste, el tromboxano A2 es un vasoconstrictor potente y agonista de la agregación plaquetaria. En la hipertensión arterial pulmonar, la síntesis de tromboxano se incrementa11.
La endotelina-1 es un vasoconstrictor que también estimula la proliferación del músculo liso de la arteria pulmonar. Los niveles plasmáticos, que se elevan en la hipertensión arterial pulmonar, tienen además una relación inversamente proporcional con el flujo de la arteria pulmonar y el gasto cardiaco12.
La síntesis del óxido nítrico, un potente vasodilatador e inhibidor de la activación plaquetaria, está catalizada por la familia de enzimas óxido nítrico sintetasa. Se han observado niveles disminuidos de la óxido nítrico sintetasa endotelial en los sujetos con hipertensión arterial pulmonar, sobre todo en aquellos con la forma idiopática. Otras moléculas relacionadas con la aparición de hipertensión arterial pulmonar incluyen la serotonina (5-hidroxitriptamina), el péptido intestinal vasoactivo, la adrenomedulina y los factores de crecimiento endotelial vascular13.
Las alteraciones metabólicas que ocurren en la hipertensión pulmonar están implicadas en la remodelación vascular y en un estado proinflamatorio. Se han propuesto anomalías mitocondriales metabólicas, por ejemplo una preferencia por la glucólisis en lugar del metabolismo oxidativo mitocondrial, lo que favorece un estado antiapoptótico característico de la hiperproliferación vascular presente en la hipertensión pulmonar, semejante a los procesos neoplásicos2.
Los factores ambientales relacionados con el aumento del riesgo para desarrollar hipertensión arterial pulmonar incluyen hipoxia, anorexígenos y estimulantes del sistema nervioso central14.
ClasificaciónLa clasificación es:
- i)
Hipertensión arterial pulmonar
- ii)
Hipertensión pulmonar debido a falla cardiaca izquierda
- iii)
Hipertensión pulmonar debido a enfermedad pulmonar y/o hipoxia
- iv)
Hipertensión pulmonar debido a enfermedad tromboembólica crónica
- v)
Hipertensión pulmonar multifactorial
Los cambios introducidos en la última clasificación del quinto simposio mundial se centraron especialmente en los pacientes del grupo 1: hipertensión arterial pulmonar. La modificación principal fue retirar la hipertensión pulmonar persistente del recién nacido del grupo 1. La hipertensión pulmonar asociada a anemia hemolítica crónica se transfirió del grupo 1 al 5 (mecanismos multifactoriales/poco claros); además, se agregaron otros términos relacionados con hipertensión pulmonar pediátrica (tabla 1)15.
Clasificación de la hipertensión pulmonar. Quinto Simposio Mundial Niza 2013
| 1. Hipertensión arterial pulmonar (PAH) |
| 1.1. Idiopática (IPAH) |
| 1.2. Familiar (FPAH) |
| 1.2.1. BMPR2 |
| 1.2.2. ALKI, endoglina, SMAD9, CAV1, KCNK3 |
| 1.2.3. Desconocido |
| 1.3. Asociada con drogas o toxinas |
| 1.4. Asociada con APAH |
| 1.4.1. Enfermedad del tejido conectivo |
| 1.4.2. Infección por VIH |
| 1.4.3. Hipertensión portal |
| 1.4.4. Enfermedad congénita cardiaca |
| 1.4.5. Esquistosomiasis |
| 1.5. Hemangiomatosis pulmonar capilar y/o enfermedad pulmonar venooclusiva |
| 1.5.1. Hipertensión pulmonar persistente del recién nacido |
| 2. Hipertensión pulmonar debido a falla cardiaca izquierda |
| 2.1. Disfunción sistólica |
| 2.2. Disfunción diastólica |
| 2.3. Enfermedad valvular |
| 2.4. Congénita/obstrucción del tracto de salida y miocardiopatías congénitas |
| 3. Hipertensión pulmonar debido a enfermedad pulmonar y/o hipoxia |
| 3.1. Enfermedad pulmonar obstructiva crónica |
| 3.2. Enfermedad intersticial del pulmón |
| 3.3. Otras enfermedades pulmonares con patrón mixto restrictivo y obstructivo |
| 3.4. Trastornos de la respiración durante el sueño |
| 3.5. Trastornos alveolares de hipoventilación |
| 3.6. Exposición crónica a grandes altitudes |
| 3.7. Anormalidades del desarrollo pulmonar |
| 4. Hipertensión pulmonar asociada a tromboembolia crónica |
| 5. Hipertensión pulmonar con mecanismos multifactoriales |
| 5.1. Enfermedades hematológicas: anemia hemolítica crónica, desórdenes mieloproliferativos, esplenectomía |
| 5.2. Enfermedades sistémicas: sarcoidosis, histiocitosis de células de Langerhans, linfangioleiomiomatosis, vasculitis, neurofibromatosis |
| 5.3. Enfermedades metabólicas: enfermedad de Gaucher, desórdenes tiroideos, enfermedades del almacenamiento del glucógeno |
| 5.4. Otras: obstrucción tumoral, mediastinitis fibrosante, enfermedad renal crónica en diálisis, HP segmentaria |
La terapia con medicamentos aprobados para hipertensión arterial pulmonar debe iniciarse en pacientes sin vasorreactividad o sin respuesta adecuada a los calcioantagonistas16.
Los antagonistas del receptor de endotelina, los inhibidores de la fosfodiesterasa tipo 5 (PDE5i) y los análogos de la prostaciclina y del óxido nítrico son eficaces y seguros para mejorar el perfil hemodinámico pulmonar, el cuadro clínico, y en algunos casos, la supervivencia en hipertensión arterial pulmonar idiopática, así como en otras formas de hipertensión arterial pulmonar2.
Los objetivos del tratamiento en los pacientes con hipertensión arterial pulmonar son múltiples e incluyen mejoría de los síntomas, calidad de vida y supervivencia. La evaluación objetiva para medir la respuesta al tratamiento incluye el progreso de la capacidad de ejercicio (prueba de marcha de 6 minutos, prueba de esfuerzo cardiopulmonar), el perfil hemodinámico y la supervivencia10.
Entre las medidas generales que deben ser implementadas figuran: dieta, ejercicio guiado, vacunas y evitar embarazos (fig. 1). Respecto al uso de nuevos anticoagulantes orales (rivaroxabán, dabigatrán y apixabán) en pacientes HTPTEC, se deben tener en cuenta las interacciones medicamentosas con ERA, PDE5i; por tanto se recomienda evitar cambios en esta terapia hasta que haya más evidencia disponible. El bosentán es un inductor del citocromo P450 3A4 (CYP3A4), que puede reducir potencialmente la concentración de los inhibidores del factor Xa, rivaroxabán y apixabán17.
Nuevos medicamentosEl tratamiento de la hipertensión arterial pulmonar ha evolucionado considerablemente en la última década debido, en parte, a los avances en el conocimiento fisiopatológico de la enfermedad y a la disponibilidad de nuevos agentes farmacológicos. Por el contrario, la HTPTEC ha recibido menos atención probablemente porque hasta el momento ha sido tratada esencialmente como una entidad quirúrgica, curable con endarterectomía pulmonar18. Sin embargo, la posibilidad de terapias farmacológicas dirigidas a la hipertensión arterial pulmonar en algunos pacientes con HTPTEC no candidatos a manejo quirúrgico, es un hecho.
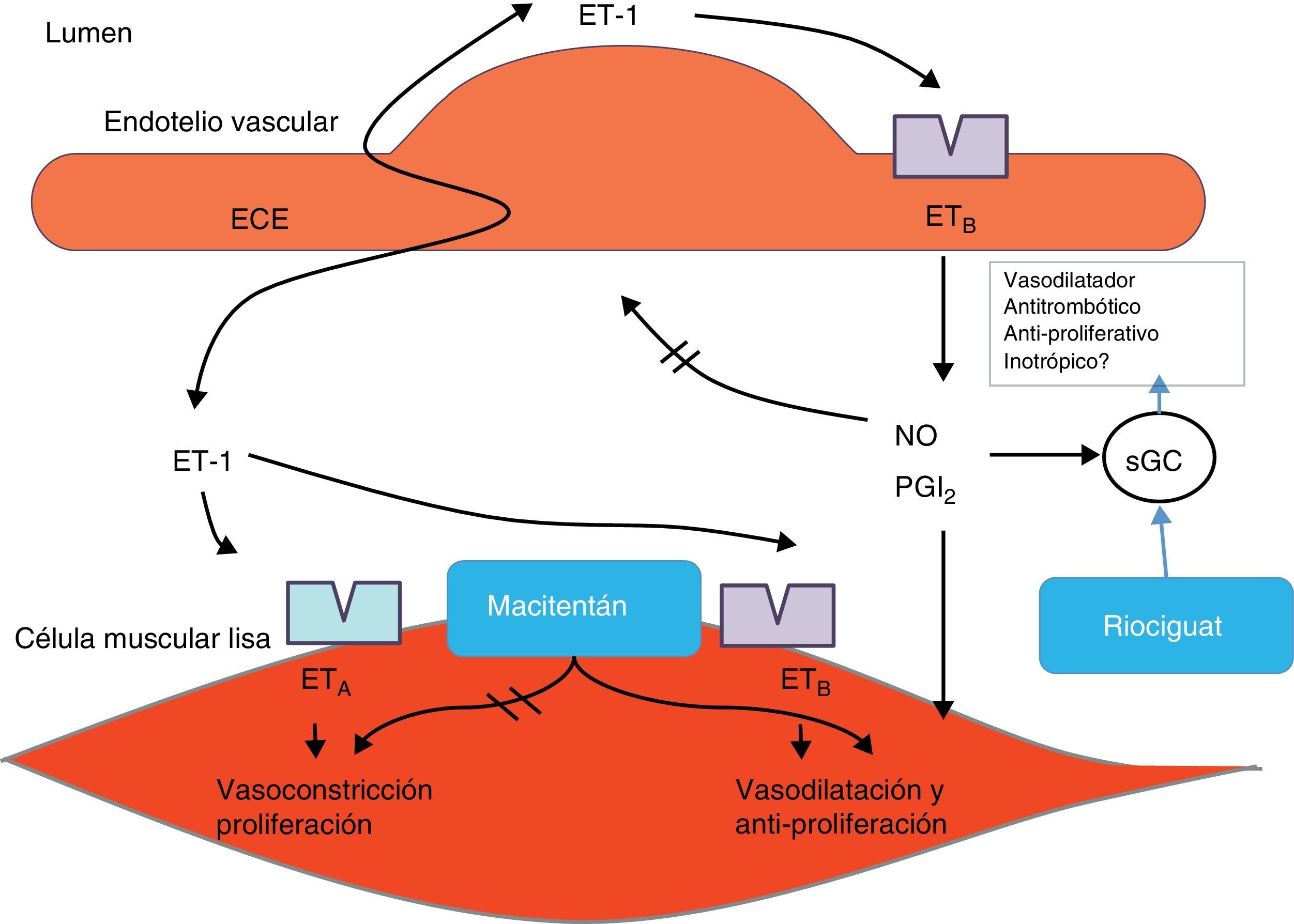
El riociguat es miembro de una nueva clase de medicamentos llamados estimuladores solubles de la guanilato ciclasa. Como se mencionó previamente, la alteración en la síntesis y en la señalización del óxido nítrico a través de la vía de la guanilato ciclasa-monofosfato, está implicada en la patogénesis de la hipertensión pulmonar. El riociguat tiene un modo de acción dual: la estimulación directa de la guanilato ciclasa soluble independiente del óxido nítrico, y el aumento de la sensibilidad de la guanilato ciclasa soluble al óxido nítrico. Este medicamento, además, aumenta el nivel de monofosfato de guanosina cíclico –GMPc–, y a su vez produce vasodilatación, así como efectos antiproliferativos y antifibróticos (fig. 2). Ghofrani et al.19 evaluaron el riociguat como tratamiento oral para dos formas de hipertensión pulmonar: HTPTEC e hipertensión arterial pulmonar (grupos 1 y 4). A continuación se discuten los hallazgos.
 es una enzima dimérica que contiene hierro y es activada por el óxido nítrico cuando está en el estado reducido (Fe2+). Riociguat es un estimulador de la sGC. Los medicamentos de esta clase aumentan la sensibilidad de la sGC en óxido nítrico y pueden estimular la forma reducida de sGC.")
Mecanismo de acción de los nuevos agentes para el manejo de la hipertensión pulmonar. La guanilato ciclasa soluble (sGC) es una enzima dimérica que contiene hierro y es activada por el óxido nítrico cuando está en el estado reducido (Fe2+). Riociguat es un estimulador de la sGC. Los medicamentos de esta clase aumentan la sensibilidad de la sGC en óxido nítrico y pueden estimular la forma reducida de sGC.
Las terapias farmacológicas han sido aprobadas para hipertensión arterial pulmonar, pero no para HTPTEC, que se caracteriza por la obstrucción de la circulación pulmonar secundaria a trombos residuales, lo cual lleva a un aumento de resistencia vascular pulmonar, hipertensión pulmonar y falla ventricular derecha. Los pacientes con HTPTEC tienen mal pronóstico si no reciben tratamiento oportuno18. El único fármaco que ha sido evaluado previamente para HTPTEC es el antagonista del receptor de endotelina: bosentán. En el estudio BENEFIT20 este disminuyó la resistencia vascular pulmonar en comparación con placebo (24%), aunque no mejoró significativamente la distancia de la caminata de seis minutos en comparación con placebo.
La investigación Chronic Thromboembolic Pulmonary Hypertension Soluble Guanylate Cyclase–Stimulator Trial 1 (CHEST-1) publicada en el New England Journal of Medicine, es un ensayo clínico fase tres, doble ciego, aleatorizado y controlado con placebo, de una duración de 16 semanas y 89 centros participantes de 26 países. En este se analizaron la eficacia y el perfil de los efectos secundarios del riociguat en pacientes entre 18 y 80 años con HTPTEC sin indicación de cirugía, o con hipertensión pulmonar persistente o recurrente después de endarterectomía pulmonar21.
En la semana 16, la distancia de la caminata de 6 minutos incrementó en promedio 39 metros en el grupo de pacientes que recibió riociguat comparado con el grupo placebo, en el que hubo una disminución de 6 metros en promedio (diferencia promedio 46 m; 95% intervalo de confianza [IC], 25 a 67; p<0,001). Mientras que en el grupo que recibió riociguat hubo una disminución significativa de 226 din/s/cm–5 en la resistencia pulmonar vascular, en el grupo placebo hubo un aumento de 23 din/s/cm–5 en esta medida (diferencia promedio de cuadrados, –246 din/s/cm; 95% IC, –303 a –190; p < 0,001). El riociguat también se asoció con mejoría significativa de las variables hemodinámicas, incluyendo PAPm y gasto cardiaco. En aquellos pacientes tratados con riociguat disminuyeron los niveles de NT-pro BNP y se observó mejoría de la clase funcional de la OMS.
Los efectos adversos graves más frecuentes fueron: falla ventricular derecha (en el 3% de los pacientes en cada grupo), síncope (en el 2% del grupo de riociguat y el 3% del grupo placebo) y hemoptisis (en el 2% del grupo riociguat). Los eventos adversos graves relacionados en el grupo riociguat incluyeron síncope (3 pacientes –2%–), gastritis, insuficiencia renal aguda e hipotensión (1%). Ninguno de estos eventos adversos graves se consideró relacionado con el fármaco en estudio.
Por otro lado, 237 pacientes (98% de los que completaron el estudio CHEST-1) entraron en el estudio de extensión CHEST-2 actualmente en curso22. De estos, 194 (129 pacientes del grupo riociguat y 65 del grupo placebo en el estudio CHEST-1) se incluyeron en el análisis que duró 336 días. El informe preliminar del estudio interino CHEST-2 mostró mejoría continuada en la distancia recorrida en la caminata de 6 minutos en el grupo que recibió riociguat. Se observó un aumento promedio (± DE) de 63 m sobre la distancia de referencia en la semana doce. Los autores demostraron que el riociguat disminuye de manera significativa los valores de la resistencia vascular pulmonar y mejora la clase funcional OMS que también se relaciona con percepción individual de beneficio y sobrevivencia.
Una ventaja del CHEST es que los autores estudiaron desenlaces esenciales para evaluar el manejo de la hipertensión arterial pulmonar. Por ejemplo, los valores hemodinámicos son mediciones importantes en los estudios de hipertensión arterial pulmonar, ya que proporcionan una medida objetiva de la circulación pulmonar y son útiles para predecir resultados. Se ha visto que si la resistencia vascular pulmonar de base supera 900 din/s/cm−5, la mortalidad se incrementa. En consecuencia, la reducción de la resistencia vascular pulmonar se asocia con mejores desenlaces y aumento de la supervivencia después de la cirugía en los pacientes con HTPTEC18.
Los investigadores concluyeron que el riociguat mejora significativamente la distancia recorrida en la caminata de 6 minutos, la resistencia vascular pulmonar y otros resultados clínicos en pacientes con HTPTEC que fueron considerados inelegibles para la cirugía o que tenían hipertensión pulmonar persistente o recurrente después de la endarterectomía pulmonar. La importancia de estos resultados radica en que actualmente el único tratamiento recomendado para pacientes con HTPTEC es la endarterectomía pulmonar. Sin embargo, únicamente el 63% tiene indicación quirúrgica; además la hipertensión arterial pulmonar persiste o recurre en 5 a 35% de los casos después de la cirugía.
Pese a las ventajas en efectividad y seguridad arrojadas en el CHEST, los beneficios de la endarterectomía pulmonar (aumento de 100 m de distancia recorrida en 6 minutos) superan los beneficios con riociguat (46 m)23, de ahí que sea preciso recordar que los pacientes que cumplen criterios para manejo quirúrgico deben recibir esta en lugar de tratamiento médico.
Riociguat para hipertensión arterial pulmonar sintomáticaGhofrani et al. condujeron el estudio PATENT-1 (Pulmonary Arterial Hypertension Soluble Guanylate Cyclase–Stimulator Trial 1)24, el cual también es fase tres, doble ciego y aleatorizado controlado con placebo. Incluyó 124 centros de 30 países y analizó la eficacia y perfil de seguridad del riociguat en pacientes con hipertensión arterial pulmonar sintomática sin tratamiento y quienes recibían antagonistas del receptor de la endotelina o prostanoides no intravenosos.
Los pacientes con hipertensión arterial pulmonar sintomática (idiopática, familiar, asociada a enfermedad del tejido conectivo, cardiopatía congénita, hipertensión portal con cirrosis hepática, consumo de anorexígenos o anfetaminas) fueron incluidos si tenían una resistencia vascular pulmonar mayor de 300 dinas/s/cm−5, PAPm al menos de 25mm Hg y caminata de 6 minutos de 150 a 450 m. La hipertensión arterial pulmonar idiopática fue el diagnóstico más común y la mayoría de los pacientes se encontraba en clase funcional ii o iii de la OMS.
En este estudio 443 pacientes fueron asignados al azar a placebo (n=126), riociguat en dosis hasta 2,5mg tres veces al día (n=254) o riociguat de 1,5mg tres veces al día (n=63).
El desenlace final primario, semejante al de los otros estudios fase tres que han estudiado la eficacia para hipertensión arterial pulmonar, fue el cambio de la línea de base hasta el final de la semana 12 en la distancia caminada en 6 minutos. En el grupo que recibió 2,5mg de riociguat, la distancia aumentó 30 m en promedio y disminuyó 6 m en el grupo placebo (diferencia 36 m, IC 95% de 20 a 52; p<0,001).
También fue evidente una diferencia significativa en los desenlaces secundarios en los pacientes tratados con riociguat. La resistencia vascular pulmonar disminuyó 223 dinas/s/cm−5 en el grupo de 2,5mg, en comparación con 9 dinas/s/cm−5 en el grupo placebo (diferencia –226 dinas/s/cm−5; IC del 95%, –281 a –170, p<0,001). También hubo mejoría importante en otras variables hemodinámicas incluyendo: presión de la arteria pulmonar media, gasto cardiaco, pro-BNP, clase funcional de la OMS y escala de disnea de Borg.
Los beneficios de la terapia riociguat se mantuvieron en el estudio de extensión de 24 semanas. Al igual que en CHEST 1, riociguat tuvo buen perfil de seguridad, pues su administración no se asoció con mayor tasa de efectos adversos graves comparado con placebo. PATENT 1 y 2 demostraron que riociguat mejoró significativamente la capacidad de ejercicio en los pacientes con hipertensión pulmonar arterial, beneficio que fue semejante en aquellos que recibían antagonistas del receptor de endotelina o prostanoides y en aquellos que no recibían tratamiento.
Una limitación del PATENT-1 es la magnitud de la efectividad alcanzada al comparar otros medicamentos orales para la hipertensión arterial pulmonar. Solo un 21% de los pacientes tratados mejoró la clase funcional OMS a las 12 semanas (en comparación con un 14% en el grupo placebo). El aumento de la distancia de caminata de 6 minutos es similar al observado en otros ensayos aleatorizados y controlados de otras terapias orales como tadalafil (PHIRST)25 y bosentán (BREATHE-1). Sin embargo, los resultados son menos impactantes que los obtenidos en el ensayo SUPER26, que demostró que sildenafil aumentó 50 m la distancia de línea de base en la caminata de seis minutos, en comparación con placebo.
No obstante, en cuanto al CHEST y al PATENT los autores no estudiaron los efectos del riociguat en el ventrículo derecho. Si bien la distancia alcanzada en la caminata de 6 minutos refleja el funcionamiento del ventrículo derecho, se debe establecer si también tiene efectos en la inotropía del mismo, que puedan relacionarse con desenlaces favorables.
Así mismo, está por definirse si los resultados expuestos en el PATENT se pueden extrapolar a otros grupos de pacientes con hipertensión pulmonar secundaria a infección por virus de inmunodeficiencia humana, esquistosomiasis o enfermedades hematológicas.
En conclusión, el riociguat está aprobado por la FDA y EMA para la hipertensión arterial pulmonar y la HPTEC.
MacitentánEs un antagonista dual del receptor de endotelina que fue desarrollado mediante la modificación de la estructura del bosentán para aumentar su eficacia y seguridad. Macitentán se caracteriza por una unión sostenida al receptor y por un aumento de la distribución tisular24. A pesar de las posibles diferencias en la actividad del receptor, la eficacia de los fármacos antagonistas de la endotelina en la hipertensión arterial pulmonar parece ser comparable.
SERAPHIN (Study Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcomes) es la investigación prospectiva más grande de hipertensión arterial pulmonar disponible hasta el momento27. Es un ensayo clínico fase tres, multicéntrico, aleatorizado, doble ciego y controlado que incluyó 742 pacientes mayores de 12 años, con hipertensión pulmonar familiar, idiopática, relacionada con enfermedad del tejido conectivo, cortocircuitos sistémico-pulmonares congénitos, infección por virus de la inmunodeficiencia humana, uso de drogas o exposición a toxinas. Otros criterios de inclusión fueron: confirmación de hipertensión arterial pulmonar por cateterismo coronario derecho, distancia mínima de 50 m en la caminata de 6 minutos y clase funcional ii, iii o iv de la OMS.
Pulido et al.27 analizaron si el tratamiento a largo plazo con macitentán redujo la morbilidad y la mortalidad en los pacientes con hipertensión arterial pulmonar en 151 centros de 39 países entre mayo de 2008 y diciembre de 2009. Los pacientes incluidos en el estudio fueron asignados de manera aleatoria en una proporción 1:1:1, para recibir placebo una vez al día, macitentán 3mg/día o macitentán 10mg/día.
El desenlace primario compuesto fue el tiempo desde el inicio del tratamiento hasta el primer evento relacionado con hipertensión arterial pulmonar (empeoramiento de la hipertensión arterial pulmonar, inicio de tratamiento con prostanoides intravenosos o subcutáneos, trasplante de pulmón o atrioseptostomía) o muerte por cualquier causa hasta el final del tratamiento. El empeoramiento de la hipertensión arterial pulmonar se definió por la presencia de tres de los siguientes parámetros: disminución de la distancia recorrida en 6 minutos de al menos un 15% del valor inicial, empeoramiento de los síntomas de hipertensión arterial pulmonar y necesidad de tratamiento adicional para esta. La exacerbación de los síntomas de hipertensión arterial pulmonar incluyó al menos uno de los siguientes criterios: cambio del valor inicial de clase funcional de la OMS (o ningún cambio en los pacientes que se encontraban en clase funcional iv de la OMS al inicio del estudio) y aparición o agudización de los síntomas de insuficiencia cardíaca derecha refractarios al manejo con diuréticos orales.
El empeoramiento de la hipertensión arterial pulmonar fue el desenlace primario más frecuente. El hazard ratio (HR) con la dosis de 3mg de macitentán versus placebo fue de 0,70 (97,5% [IC]: 0,52 a 0,96; p=0,01), mientras que en el grupo que recibió 10mg de macitentán versus placebo fue de 0,55 (IC del 97,5%, 0,39 a 0,76, p<0,001).
El macitentán redujo significativamente la morbimortalidad en los sujetos con hipertensión arterial pulmonar. El desenlace compuesto de muerte por hipertensión arterial pulmonar u hospitalización por hipertensión arterial pulmonar ocurrió en 84 pacientes (33,6%) con el placebo, en 65 (26,0%) con 3mg de macitentán y en 50 (20,7%) con 10mg de macitentán. El HR en el grupo que recibió 3mg de macitentán versus placebo fue de 0,67 (IC del 97,5%; 0,46-0,97, p=0,01) y el HR en el grupo de 10mg de macitentán versus placebo fue 0,50 (IC del 97,5%; 0,34-0,75, p<0,001). Cuando se analizó la mortalidad independiente de la causa hubo una tendencia hacia la reducción en el grupo que recibió 10mg de macitentán comparado con placebo. Sin embargo, el estudio no obtuvo poder suficiente para demostrar un efecto en la mortalidad como único desenlace.
La clase funcional mejoró desde el inicio hasta 6 meses en el 13% de los pacientes en el grupo de placebo, en comparación con el 20% del grupo que recibió 3mg de macitentán (p=0,04) y 22% del grupo que recibió 10mg de macitentán (p=0,006). Los pacientes que recibieron macitentán independiente de la dosis, también presentaron disminución significativa en los valores de resistencia vascular pulmonar y aumento importante en los índices cardiacos comparado con el grupo placebo.
Los eventos secundarios más comunes en todos los grupos comparados fueron elevación de transaminasas y edema periférico. Los pacientes que recibieron tratamiento con macitentán reportaron mayor frecuencia de nasofaringitis, cefalea y anemia.
Además de tratarse de una investigación con alto rigor metodológico, otra ventaja de SERAPHIN es que a diferencia de otros estudios incluyó múltiples desenlaces clínicos. Además, la duración del estudio (en promedio en el grupo de tratamiento con 3mg de macitentán 99,5 semanas y 103,9 semanas en el grupo que recibió 10mg de macitentán) fue significativamente mayor comparada con la duración de otros estudios clínicos.
El macitentán ha sido aprobado por la FDA y la EMA para el manejo de la hipertensión arterial pulmonar.
Nuevas terapias en desarrolloUna mayor comprensión de la epidemiología, patogenia y fisiopatología de la hipertensión arterial pulmonar ha dado lugar a avances significativos en el campo terapéutico; sin embargo la enfermedad sigue siendo mortal. Las opciones de tratamiento no están disponibles de forma universal y no siempre son eficaces, hecho que enfatiza la necesidad de desarrollar nuevas terapias y estrategias terapéuticas. En este sentido, la búsqueda de genes, así como la terapia génica y con células madre continúan siendo objetivos clave.
Por su parte, imatinib, inhibidor de la tirosín-kinasa y del factor de crecimiento derivado de plaquetas, demostró mejorar el perfil hemodinámico, aunque produjo serios efectos adversos28. Terguride, medicamento que modula los niveles de neurotransmisores al bloquear el receptor tipo 2 de serotonina (5-hidroxitriptamine) redujo la progresión de hipertensión arterial pulmonar en ratas, pero los resultados de fase 2 fueron negativos29. También hay un creciente interés en avipath, una molécula sintética de péptido intestinal vasoactivo, y en selexipag, un agonista no prostanoide del receptor de prostaciclina30.
ConclusionesSi bien los resultados de los estudios discutidos previamente son muy alentadores, aún es necesario abordar la eficacia y seguridad de riociguat y macitentán en comparación con otras terapias orales aprobadas para la hipertensión arterial pulmonar. Se debe tener presente que la decisión para iniciar tratamiento farmacológico óptimo debe ser individualizada y tener en cuenta distintos factores como la severidad de la enfermedad, la ruta de administración, los efectos adversos, las comorbilidades y los costos de la terapia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A Ana Cristina Montenegro.