




La artritis reumatoide es una enfermedad crónica, sistémica e inflamatoria, de etiología desconocida, que afecta, principalmente la membrana sinovial de las articulaciones. Se caracteriza por dolor crónico, y destrucción articular que conlleva un aumento de la mortalidad y un elevado riesgo de invalidez con altos costos para el enfermo y la sociedad. Describimos el caso de un paciente joven con anemia aplásica desde los 8 años, que inicia en 2009 cuadro de dolor y aumento de volumen articular en manos, muñecas y tobillos bilaterales, cumpliendo los criterios para artritis reumatoide.
Rheumatoid arthritis (RA) is a chronic, systemic, and inflammatory disease of unknown etiology that mainly affects the synovial membrane of the joints. It is characterized by chronic pain and joint destruction, which leads to premature mortality and risk of disability, with high costs to the patient and society. The case is presented of a young male patient with aplastic anemia since 8 years old who, in 2009, began with swelling and joint pain in the hands, wrists and ankles, fulfilling criteria for rheumatoid arthritis.
La artritis reumatoide (AR) es una enfermedad crónica, sistémica e inf lamatoria, de etiología desconocida, que afecta, principalmente la membrana sinovial de las articulaciones, con una prevalencia estimada de 0,5–1% de la población mundial, afectando, en especial, a mujeres entre los 30 y 50 años1.
El inicio de la enfermedad suele ser insidioso, con los síntomas predominantes de dolor, rigidez y edema articular. Por lo general, las articulaciones interfalángicas proximales, metacarpofalángicas y metatarsofalángicas de las manos y pies son sitios de artritis precoz en la enfermedad. Otras articulaciones de los miembros superiores e inferiores, como los codos, hombros, tobillos y rodillas, también son comúnmente afectadas2.
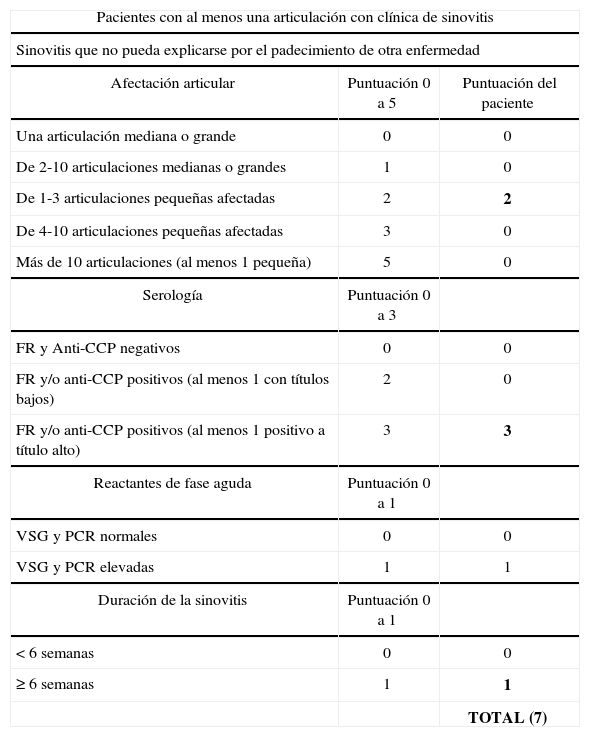
Dentro de los criterios diagnósticos formulados en 2010, por el Colegio Americano de Reumatología y la Liga Europea contra el Reumatismo para el diagnóstico de AR, se incluye por lo menos una articulación con sinovitis clínica y que esta no pueda explicarse por el padecimiento de otra enfermedad. Un paciente será clasificado con AR si la suma total es ≥ 6, puntuación obtenida en cada uno de los 4 dominios. Los dominios establecidos son el número y localización de las articulaciones afectadas (puntuación de 0 a 5), alteraciones serológicas (puntuación de 0 a 3), elevación de los reactantes de fase aguda (puntuación de 0 a 1) y duración de los síntomas (puntuación de 0 a 1)3.
Las características extraarticulares de la AR incluyen anemia de tipo inflamatoria por enfermedad crónica, fatiga, nódulos, pleuropericarditis, neuropatía, epiescleritis, escleritis, esplenomegalia, vasculitis y enfermedad renal, que pueden ocurrir durante el curso de la enfermedad4.
El uso temprano de medicamentos como antiinflamatorios no esteroideos (AINE), corticosteroides, drogas modificadoras de la enfermedad (metotrexato, leflunomida, hidroxicloroquina, sulfasalazina) e inhibidores del factor de necrosis tumoral puede cambiar de forma significativa el curso clínico de la AR, evitando así el daño ar ticular y la discapacidad5.
Presentamos el caso de un paciente joven de sexo masculino, con historia de anemia aplásica (AA) desde los 8 años, que manifiesta AR infrecuente para el género y la edad. Lo relevante de este caso es la asociación de la manifestación hematológica con la AR, entidades que no coexisten habitualmente, permitiéndonos hacer una revisión bibliográfica y reportar esta asociación clínica como la primera en nuestro medio.
Presentación del casoPaciente de 24 años, sexo masculino, blanco, natural de Río de Janeiro, con diagnóstico de AA desde los 8 años, que inicia con sinusitis repetitiva, epistaxis, astenia y adinamia, sin esplenomegalia o linfoadenopatías; los exámenes de laboratorio demostraban un síndrome anémico macrocítico con una hemoglobina que oscilaba entre 7 y 9mg/dl, hematocritos entre 27–30%, conteo de neutrófilos absolutos entre 1.300 y 1.500/mm3, plaquetas: 28.000 a 90.000/mm3, reticulocitos: < 1%, la capacidad de fijación del hierro no estaba elevada, los niveles de ferritina sérica y de receptores séricos de transferrina estaban normales, niveles de folatos y vitamina B12 normales, fue realizado un aspirado de médula ósea con pocas células hematopoyéticas, y una biopsia de médula ósea compatible con AA.
El paciente fue tratado con múltiples hemotransfusiones y prednisona 10mg/día, con poca respuesta clínica y sin mejoría de los parámetros de laboratorio, siendo adicionados ciclosporina 100mg/día y ácido fólico 5mg/día con estabilización del cuadro clínico.
En 2009, inicia cuadro de dolor y aumento de volumen articular, en articulaciones interfalángicas proximales, muñecas y tobillos bilaterales, de inicio súbito, sin historia de trauma u otro antecedente relevante, persistente por más de 6 meses y acompañado de fatiga, rigidez matinal > 1h, con limitación para la flexión y extensión de la muñeca y desviación lateral de los tobillos.
En el examen físico, el paciente presentaba edema, calor y dolor a la compresión lateral de la segunda a la quinta metacarpofalángica derecha e izquierda, y segunda, tercera y cuarta interfalángica proximal izquierda (test de Squeeze positivo). En muñecas y tobillos bilaterales presentaba edema, calor y dolor a la palpación de la articulación, sin entesitis.
Los exámenes de laboratorio están descritos en la tabla 1. La radiografía de muñeca, manos y dedos demostraba edema de tejidos blandos, con disminución de la densidad ósea y erosiones en tercera y cuarta interfalángica proximal izquierda. Las radiografías de pies y tobillos presentaban disminución de la densidad ósea, sin erosiones.
Resultados de laboratorio clínico del paciente
| HB: 9mg/dl | Colesterol: 170mg/dl |
| HCTO: 30% | Triglicéridos: 125mg/dl |
| VCM: 87fL | FR: 158U/ml (VR: < 20U/ml) Nefelometría |
| HCM: 25 pg | Anti-CCP: 175U/L (VR: < 20U/L) |
| Leucocitos: 7.000/mm3 | C3: 134mg/dl (VR: 88-201mg/dl) |
| Neutrófilos: 60% | C4: 38mg/dl (VR: 16-47mg/dl) |
| Linfocitos: 40% | Anti-VIH: negativo |
| Plaquetas: 120.000/mm3 | Anti-SSA/Ro: < 1,0 (VR: > 12,5U/mL) negativo |
| VSG: 96mm/h | Anti-SSB/La: negativo |
| PCR: 131mg/L (VR: < 3mg/L) | Anti-Sm: negativo |
| GOT: 35U/L (VR: 10-40U/L) | Anti-ADN: negativo |
| GPT: 37U/L (VR: 7-40U/L) | Anti-RNP: negativo |
| GGT: 37U/L (VR: 6-50U/L) | HBs-Ag: negativo |
| Creatinina: 0,9mg/dl | Anti-HBs: 10U/ml (VR: < 10U/ml) |
| Urea: 22mg/dl | Anti-HBc: negativo |
| Glucosa: 87mg/dl | Anti-VHC: negativo |
FR: factor reumatoide; HB: hemoglobina; HTCO: hematocritos; VCM: volumen corpuscular medio; VR: valor de referencia; VSG: velocidad de sedimentación globular.
El paciente fue diagnosticado de AR al presentar una puntuación de 7 en los criterios formulados por Colegio Americano de Reumatología/Liga Europea contra el Reumatismo 2010 (tabla 2). Fue iniciado tratamiento con metotrexato 12,5mg/semana y prednisona 7,5mg/día.
Criterios diagnósticos del paciente formulados por Colegio Americano de Reumatología/Liga Europea contra el Reumatismo 2010
| Pacientes con al menos una articulación con clínica de sinovitis | ||
| Sinovitis que no pueda explicarse por el padecimiento de otra enfermedad | ||
| Afectación articular | Puntuación 0 a 5 | Puntuación del paciente |
| Una articulación mediana o grande | 0 | 0 |
| De 2-10 articulaciones medianas o grandes | 1 | 0 |
| De 1-3 articulaciones pequeñas afectadas | 2 | 2 |
| De 4-10 articulaciones pequeñas afectadas | 3 | 0 |
| Más de 10 articulaciones (al menos 1 pequeña) | 5 | 0 |
| Serología | Puntuación 0 a 3 | |
| FR y Anti-CCP negativos | 0 | 0 |
| FR y/o anti-CCP positivos (al menos 1 con títulos bajos) | 2 | 0 |
| FR y/o anti-CCP positivos (al menos 1 positivo a título alto) | 3 | 3 |
| Reactantes de fase aguda | Puntuación 0 a 1 | |
| VSG y PCR normales | 0 | 0 |
| VSG y PCR elevadas | 1 | 1 |
| Duración de la sinovitis | Puntuación 0 a 1 | |
| < 6 semanas | 0 | 0 |
| ≥ 6 semanas | 1 | 1 |
| TOTAL (7) | ||
FR: factor reumatoide; PCR: proteína C reactiva; VSG: velocidad de sedimentación globular.
Después de 6 meses de tratamiento, no se observó mejoría significativa y el paciente persistía con artritis de manos bilateral, que afectaba a todas las interfalanges proximales con artritis de muñeca derecha y bloqueo articular, el DAS28: 5,54 demostraba alta actividad, por lo que se aumentó el metotrexato a 17,5mg/semana, y la prednisona a 12,5mg/día.
Después de 8 meses de mantener el esquema terapéutico propuesto, el paciente presentaba discreta mejoría. La radiografía de manos y muñecas mostraba aumento de partes blandas, desmineralización periarticular y erosiones en muñeca derecha y en algunas articulaciones interfalángicas proximales bilaterales.
Los exámenes de laboratorio reportaron hematocrito: 27%, hemoglobina: 9,2g/dl, volumen corpuscular medio: 85 fL, hemoglobina corpuscular media: 26 pg, leucocitos: 8.500/mm3, plaquetas: 150.000/mm3, velocidad de sedimentación globular: 54mm/h, proteína C reactiva: 32mg/L (VR: < 3mg/L), transaminasa glutámico-oxalacética: 139U/L (VR: 10–40U/L), transaminasa glutámico-pirúvica: 141U/L (VR: 7–40U/L), gammaglutamil transpeptidasa: 145U/L (VR: 6–50U/L). Factor reumatoide: 1/160 (látex), anticuerpos antinucleares y anticuerpos extractables del núcleo: negativos (técnica ELISA).
Debido a la poca respuesta clínica con corticosteroides y drogas modificadoras de la enfermedad, como el metotrexato, y teniendo como limitante el uso de AINE, hidroxicloroquina, sulfasalasina y leflunomida, medicamentos inductores de discrasia sanguínea incluyendo agranulocitosis, AA y trombocitopenia, fue indicado como tratamiento antifactor de necrosis tumoral etarnecept 25mg, 2 veces por semana (precedido de radiografía de tórax: normal y derivado proteico purificado negativo); fue suspendido el metotrexato por elevación de las enzimas hepáticas, presentando mejoría importante de la sintomatología clínica con disminución del DAS28 a 3,6.
DiscusiónLa AR es una enfermedad autoinmune, inflamatoria, crónica y sistémica, de etiología desconocida. La artritis, generalmente es simétrica, puede conducir a la destrucción de las articulaciones debido a la erosión del cartílago y del hueso subcondral, y en más del 80% de los pacientes que tienen menos de 2 años de enfermedad hay evidencia radiológica de erosiones. Cerca del 60% de los pacientes cursa con anemia propia de las enfermedades crónicas de carácter inflamatorio6,7.
La AA se caracteriza por disminución o ausencia de los precursores hematopoyéticos de la médula ósea, por lo general, debido a una lesión en la célula madre pluripotencial8. Esta entidad puede ser congénita como la anemia de Fanconi o de tipo adquirido, como el caso del paciente, en la cual debe ser considerada la presencia de pancitopenia (anemia, trombocitopenia y neutropenia) para realizar el diagnóstico9.
Al hacer la revisión de la literatura médica, encontramos la asociación de AA con otras enfermedades autoinmunes diferentes a la AR, que generalmente se presenta en pacientes de mayor edad, de sexo femenino y después del uso de medicación mielotóxica. Pagani et al. describen 2 casos, el primero de ellos de una paciente femenina de 45 años con diagnóstico previo de síndrome de Sjögren y lupus discoide que presenta una aplasia eritroide pura, y el segundo caso de una paciente femenina de 49 años, con diagnóstico de aplasia mieloide pura, que después de 7 años presenta síndrome de Sjögren10.
Mavragani et al. describen otro caso de una mujer joven con sobreposición de síndrome de Sjögren y lupus eritematoso sistémico que presenta como complicación AA11.
Matsumoto et al. reportan el caso de una paciente femenina de 47 años con AA como complicación del síndrome de Sjögren12.
La literatura médica demuestra que la AA, generalmente está asociada a otros trastornos sanguíneos como la hemo globinuria paroxística nocturna, siendo pocos los casos relacionados con enfermedades autoinmunes13.
Por otro lado, medicamentos que son utilizados para el tratamiento de la AR, como los AINE, hidroxicloroquina, sulfasalazina, D-penicilamina y sales de oro, han estado implicados en el desarrollo de AA, como lo demuestra un estudio realizado por Nara N14.
Otra investigación realizada por Wüsthof et al. describe la presencia de AA severa con el uso de leflunomida, medicamento utilizado como modificador de la enfermedad en la AR15.
La mayoría de los episodios de agranulocitosis, que se producen entre el 1 y el 2% de los pacientes tratados con sulfasalazina, son leves, transitorios y, generalmente, surgen en los 3 primeros meses de tratamiento16. Estudios realizados por Youssef et al. demuestran que la sulfasalazina, además de producir agranulocitosis, está fuertemente implicada en el desarrollo de la AA17.
En el caso descrito, se presentó dificultad para el control de la AR inicial, debido a la limitación en el uso de AINE y algunas drogas modificadoras de la enfermedad, como hidroxicloroquina, sulfasalazina y lef lunomida, implicadas en la etiología de la AA. El paciente respondió bien al tratamiento con el medicamento biológico antifactor de necrosis tumoral alfa (etarnecept) para control de la AR, presentando mejoría importante de la sintomatología clínica con disminución del DAS28 de 5,54 a 3,6.
Descartamos la posibilidad de que algunas de estas drogas hayan sido implicadas como agentes etiológicos de la AA, debido a que el paciente presentó la AR años después de la manifestación hematológica.
Luego de una búsqueda en PubMed, Cochrane (palabras clave AA y artritis reumatoide), con fecha de corte 21 de abril de 2013, no encontramos reportes donde la AR esté asociada a la AA, lo que constituye un cuadro clínico único reportado hasta el momento en la literatura.
