



Las vasculitis sistémicas idiopáticas se caracterizan por inflamación y necrosis de las paredes de los vasos de origen desconocido1; se han propuesto medicamentos e infecciones como posibles disparadores de estas enfermedades2. A continuación se presenta el caso de un paciente con diagnóstico de granulomatosis con poliangeítis, con anticuerpos contra el citoplasma de neutrófilos tanto antiproteinasa 3 como antimieloperoxidasa y con el hallazgo poco usual de granuloma renal.
Idiopathic systemic vasculitis is characterized by inflammation and necrosis of the vessel walls of unknown origin. Medications and infections have been proposed as potential triggers of these diseases. The case is presented on a patient diagnosed with granulomatosis with polyangiitis with antibodies to neutrophil cytoplasm, as well as anti-proteinase 3 and anti-myeloperoxidase, plus the unusual finding of renal granuloma.
Hombre de 29 años, previamente sano, sin antecedente de consumo de tabaco ni otros tóxicos ni de contacto con tuberculosis, que se presentó con cuadro clínico de 2 años de evolución de tos con expectoración purulenta, ocasionalmente hemoptoica, disnea y sibilancias, para lo cual recibió tratamiento con broncodilatadores con mejoría parcial, pero con posterior aparición de fiebre hasta 39°C, sin patrón específico, pérdida de 20kg de peso, un episodio de melenas, parestesias de distribución en guante y calcetín, con disminución de la fuerza muscular distal y aparición de úlceras necróticas en tobillos y pies.
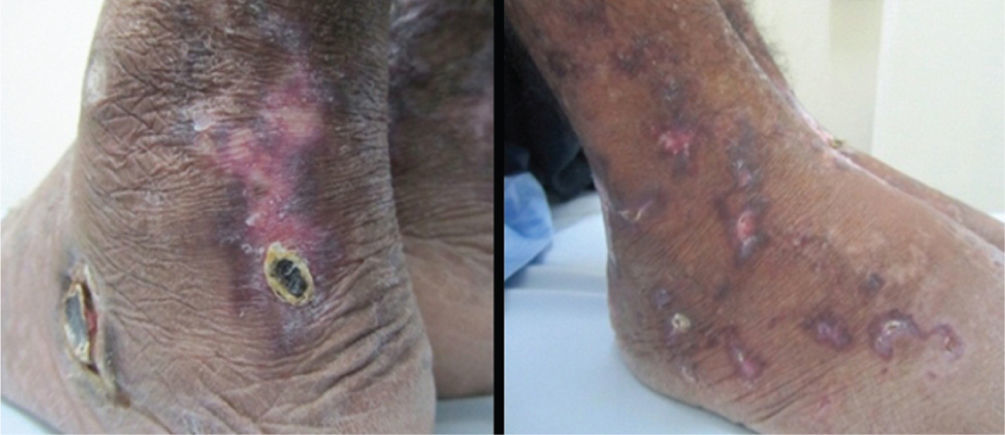
En el examen físico se encontraba caquéctico, febril, taquicárdico, normotenso, con ganglios palpables generalizados menores de 1cm, disminución de los ruidos respiratorios de forma global, sin dolor a la palpación abdominal ni hepatoesplenomegalia. En tobillos y pies tenía úlceras de aspecto necrótico, con bordes violáceos bien delimitados, algunas con costra superficial y otras que habían cicatrizado dejando un área central atrófica e hipocrómica. La fuerza muscular se encontraba disminuida distalmente (3/5 en miembros superiores y 4/5 en miembros inferiores), con hipoestesia y disestesias hasta los codos y muslos e hiporreflexia aquiliana.
En los exámenes de laboratorio se encontró elevación de los reactantes de fase aguda (leucocitosis hasta 24.000células/mm3 con neutrofilia de 21.000células/mm3, trombocitosis hasta 645.000células/mm3, eritrosedimentación de 120mm/h y proteína C reactiva hasta 9,38mg/dl), anemia microcítica hipocrómica (hemoglobina hasta 6,8g/dl con volumen corpuscular medio de 80,8) con ferritina de 870ng/dl y niveles bajos de hierro sérico. Tenía hematuria microscópica y proteinuria de 986mg/24h con depuración de creatinina de 142ml/min.
Los anticuerpos contra el citoplasma de neutrofilos (ANCA) por ELISA fueron positivos tanto para antiproteinasa 3 (PR3)-ANCA como para antimieloperoxidasa (MPO)-ANCA (39,1U/ml y 38,9U/ml, respectivamente, con valor normal < 5). Los anticuerpos antinucleares, anticuerpos contra los antígenos extractables del núcleo, el anti-ADN, ELISA para virus de la inmunodeficiencia humana, la serología para hepatotropos y la prueba serológica para la sífilis fueron negativos.
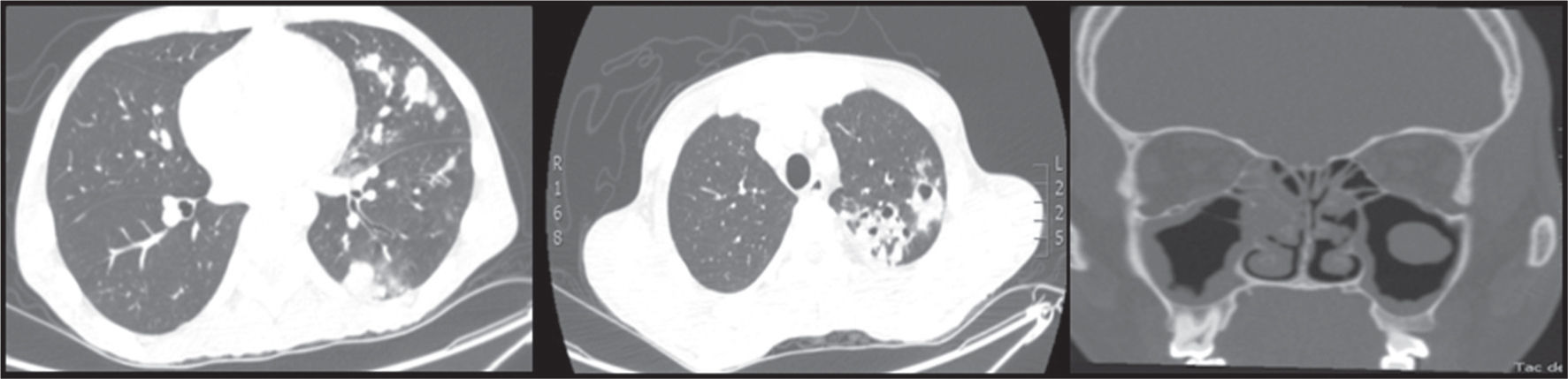
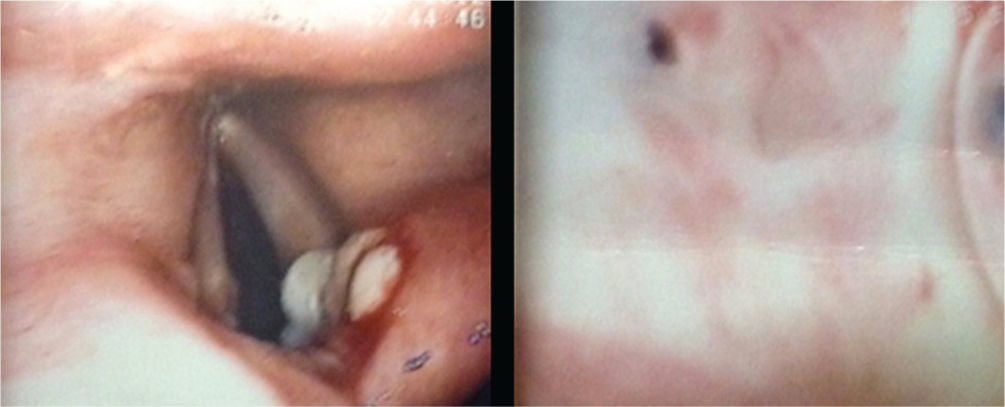
La tomografía de alta resolución de tórax demostró consolidación de la língula, nódulos centrolobulillares, signo del halo con patrón de árbol en gemación y nódulos cavitados hacia el vértice pulmonar izquierdo y escasos nódulos sólidos en lóbulo superior derecho (fig. 1 izquierda y centro). La tomografía de senos paranasales mostró pansinusitis (fig. 1 derecha). Se realizó fibrobroncoscopia, observándose en la comisura posterior del pliegue vocal derecho una lesión blanquecina de base verrugosa, cuya histología mostró inflamación aguda sin granulomas (fig. 2 izquierda), además, una estenosis de tipo diafragma en el subsegmento del segmento posterior del lóbulo posterior izquierdo (fig. 2 derecha). Las coloraciones de Ziehl Nielsen y la proteína C reactiva para Mycobacterium tuberculosis fueron negativas en esputo y en lavado broncoalveolar. Se demostró colonización por Staphylococcus aureus resistente a oxacilina en tracto respiratorio superior, pulmón y oído medio.
 y tomografía de senos paranasales (derecha).")

La electromiografía y conducciones nerviosas confirmaron polineuropatía sensitivo motora axonal con denervación. La endoscopia de vías digestivas superiores demostró la presencia de una úlcera gástrica activa sin que se reportara en la histología malignidad ni vasculitis.
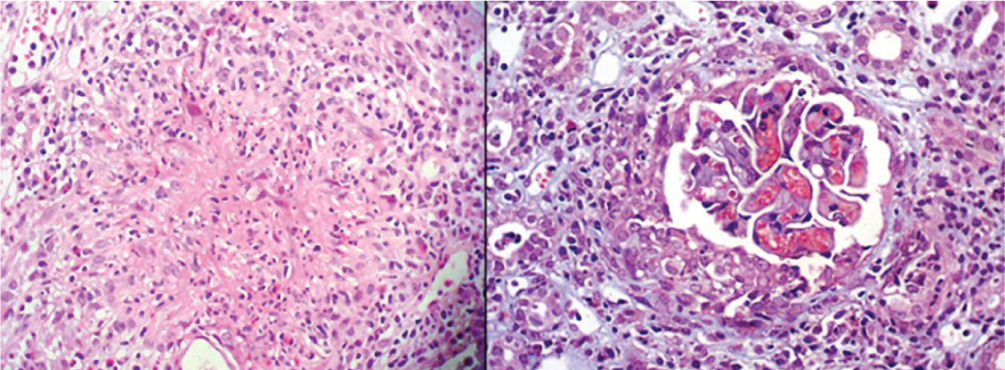
La biopsia de piel no fue concluyente pero en la biopsia renal se confirmó la presencia de vasculitis necrosante de pequeños vasos con granuloma sin eosinófilos (fig. 3), ratificando la sospecha diagnóstica de vasculitis asociada a ANCA tipo granulomatosis con poliangeítis (PG) (antigua granulomatosis de Wegener).
. Derecha: semiluna y necrosis (tricrómico 400×).")
Durante la evolución, tuvo otitis media aguda por Staphylococcus aureus resistente a meticilina para lo que recibió tratamiento antibiótico por 7 días con resolución. Con el inició del protocolo de la Sociedad Europea de Vasculitis con esteroides en dosis altas y ciclofosfamida venosa3, el paciente tuvo resolución del síndrome constitucional, disminución del tamaño de los nódulos y cavitaciones pulmonares, mejoría sintomática de la polineuropatía y cicatrización casi completa de las úlceras (fig. 4).
Discusión
Las vasculitis asociadas a ANCA incluyen la GP, la poliangeítis microscópica (PAM), granulomatosis eosinofílica con poliangeítis y la vasculitis pauci-inmune limitada al riñón4. Su diagnóstico es complejo dado que sus manifestaciones son muy diversas; el diagnóstico diferencial es amplio (otras enfermedades autoinmunes, infecciones por virus, hongos y micobacterias, neoplasias y reacciones a medicamentos)5 y los criterios de clasificación son de poca utilidad para el diagnóstico temprano6,7.
Generalmente se presentan con síntomas multisistémicos8, el pico de incidencia oscila entre los 65 y 74 años, pero varía según la población7,9–14, y tienen un gradiente de incidencia relacionado con la latitud, pues al parecer es más común en Europa y en pacientes de raza blanca15. Se ha identificado un aumento en su incidencia16 y una mayor prevalencia17 probablemente relacionadas con una mayor detección de las mismas.
Hasta el momento, su patogénesis no es clara, pero se han propuesto múltiples vías que no son excluyentes entre sí. Hay una importante predisposición genética18 con la participación variable de autoanticuerpos (ANCA y otros dirigidos contra células endoteliales)19,20, de macrófagos y neutrófilos21–24, del complemento25 y alteración en la producción de diferentes citocinas (aumento de interleucina-17 e interleucina-1β)26–29.
La detección en suero de los ANCA es una herramienta diagnóstica valiosa, sin embargo, su positividad varía, siendo del 80–90% en los casos de GP y PAM y solamente del 50% en la granulomatosis eosinofílica con poliangeítis. Cuando se combina la detección por inmunofluorescencia indirecta y por ELISA, la sensibilidad de esta prueba diagnóstica es del 80% con una especificidad del 95%30. Los pacientes con GP, generalmente son positivos para el tipo c-ANCA, específicamente PR3-ANCA31; sin embargo, el 16,7% de los pacientes tienen MPO-ANCA y hasta en el 14,8% de pacientes, los ANCA son indeterminados32 y podrían estar dirigidos contra la elastasa o la lactoferrina33. El tipo de ANCA tiene valor pronóstico pues se ha descrito que los pacientes PR3-ANCA positivos tienen una mayor probabilidad de presentar recaídas de su enfermedad durante el tratamiento34.
La doble positividad de PR-3 ANCA y MPO-ANCA en el mismo paciente es un evento poco común, describiéndose hasta en el 11% de pacientes con glomerulonefritis rápidamente progresiva, de una serie japonesa35.
En una serie de 173 pacientes con PAM o GP y compromiso renal, solo 3 pacientes tenían doble positividad, 2 de ellos tenían c-ANCA por inmunofluorescencia indirecta y 1 no tenía reporte del inmunofluorescencia indirecta36. En una serie de pacientes chinos, de 426 pacientes con vasculitis asociada a ANCA, un total de 10 pacientes era anti-PR3 y anti-MPO positivos, de estos, 1 tenía c-ANCA, 8p-ANCA, y 1 c-ANCA/p-ANCA positivos37.
En una serie holandesa con suero de 275 pacientes con vasculitis, 184 controles con otras enfermedades autoinmunes y 740 controles sanos (total 1.204), 100 eran positivos tanto para anti-PR3 como para anti-MPO30. Luego de reexaminar las muestras, en solo 10 de ellos persistía la doble positividad, 7 con predominio de anti-MPO y bajos títulos anti-PR3, 2 tenían niveles elevados de anti-PR3 y bajos de anti-MPO, solo 1 era fuertemente positivo para ambos. Dos pacientes tenían GP (uno c-ANCA y el otro p-ANCA), 5PAM, 1 poliarteritis nodosa, 1 glomerulonefritis rápidamente progresiva idiopática y 1 lupus eritematoso sistémico30.
Sin embargo, la doble positividad para PR3-ANCA y MPO-ANCA no es exclusiva de las enfermedades autoinmunes, aun más, se ha descrito una mayor frecuencia en algunas infecciones, llegando hasta un 35% de los casos38 (siendo las más comunes endocarditis subaguda, infecciones por M. tuberculosis y hepatitis C). Igualmente, la doble positividad se ha descrito en casos de vasculitis inducida por medicamentos2. Cuando se sospecha que la condición subyacente asociada a los ANCA positivos no es una vasculitis primaria algunos elementos que podrían ayudar en su diferenciación serían: hipocomplementemia, crioglobulinemia y la positividad para otros anticuerpos (como anticardiolipinas, anti-B2GPI), que sugieren la presencia de una infección crónica subyacente38.
Un hallazgo poco habitual encontrado en este paciente es la evidencia de la reacción granulomatosa en la biopsia renal, evidente solamente en el 2–9% de los casos de GP, pues la descripción más común es la de glomerulonefritis necrosante con semilunas en el 65% de los casos36, y la reacción granulomatosa, por su parte, es evidente en el 2–9% de los casos de granulomatosis con GP36,37.
El tratamiento de esta enfermedad8,39,40 y el de sus posibles diagnósticos diferenciales son muy diferentes y sería catastrófico administrar la inmunosupresión requerida en una vasculitis sistémica en pacientes con una infección de base. Se requiere de un alto grado de certeza, que muchas veces no se tiene, para el inicio de medicamentos.
En el caso presentado, se consideró el diagnóstico de granulomatosis con GP por la forma de presentación con manifestaciones multisistémicas, síndrome constitucional, presencia de infiltrados y nódulos cavitados pulmonares, pansinusitis, lesión inflamatoria de aspecto verrugoso en pliegues vocales con estenosis bronquial, afección renal con hematuria y proteinuria en rango no nefrótico, polineuropatía, úlceras en miembros inferiores, elevación de reactantes de fase aguda, ANCA positivos y exclusión de otras causas; se contó con una gran ventaja adicional que fue el hallazgo de la histopatología característica en la biopsia renal de vasculitis necrosante de pequeños vasos con granuloma sin eosinófilos, lo que constituyó la confirmación diagnóstica en este paciente con doble positividad para los ANCA.
ConclusiónEl hallazgo de granulomas en la biopsia renal y la doble positividad para anti-PR3 y anti-MPO en la GP son eventos poco comunes, el primero orientando hacia el diagnóstico pero el segundo actuando como un factor de confusión. El manejo adecuado de estas patologías es un gran reto para el clínico debido a la diversidad de manifestaciones clínicas y al amplio diagnóstico diferencial.
Responsabilidades éticasProtección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no presentar ningún conflicto de intereses en el momento de la redacción del manuscrito.
