



La neumonía intersticial linfocítica es una complicación, poco frecuente, asociada con el lupus eritematoso sistémico, sin embargo, con gran impacto en la calidad de vida. Se asocia con la presencia de anti Ro/SSA, anti La/SSB y con el diagnóstico de síndrome de Sjögren secundario. No es clara la estrategia terapéutica y la información existente está basada en reportes de caso sin disponibilidad de estudios adecuadamente diseñados. En el presente documento se expone el caso de una paciente con lupus eritematoso sistémico y síndrome de Sjögren secundario, que presentó una enfermedad pulmonar intersticial con características de neumonía intersticial linfocítica.
Lymphocytic interstitial pneumonia is an infrequent complication associated with systemic lupus erythematosus and has a great impact on quality of life. It is associated with the presence of anti-Ro/SSA, anti-La/SSB, and the diagnosis of secondary Sjögren Syndrome. Its therapeutic strategy is not clear, and the existing information is based on case reports, with there being no properly designed studies available. The case is presented here of a patient with systemic lupus erythematosus and secondary Sjögren's syndrome, who also presented with interstitial lung disease with characteristics of lymphocytic interstitial pneumonia.
El lupus eritematoso sistémico (LES) es una enfermedad autoinmunitaria con una multitud de síntomas y signos, afecta varios órganos y se asocia con una gran variedad de hallazgos inmunológicos y de laboratorio. A pesar de que la supervivencia de estos pacientes se ha incrementado notablemente en las últimas seis décadas, a más del 90% a los 5 años1, el daño de uno o varios órganos sigue generando una gran morbilidad y, en algunas ocasiones, se convierte en todo un reto diagnóstico para el clínico. El parénquima pulmonar habitualmente pasa desapercibido en las afecciones autoinmunitarias, excepción hecha en casos puntuales como la esclerosis sistémica, siendo más llamativo el compromiso vascular pulmonar en estas enfermedades, sin embargo, la morbilidad generada por procesos inflamatorios agudos y crónicos en el caso del LES o en el caso del compromiso intersticial en el síndrome (Sd) de Sjögren pueden conllevar una significativa secuela funcional. Exponemos el caso de una paciente con LES y Sd de Sjögren secundario (SSs) a quien se le identificó clínica, radiológica e histológicamente la presencia de neumonía intersticial linfocítica (NIL) y se realiza una revisión narrativa de la literatura.
CasoPaciente de sexo femenino, de 61 años de edad, con diagnóstico de LES desde el 2002, que debuta con poliserositis (pleuritis, derrame pleural, pericarditis y líquido libre cavidad peritoneal), artritis de grandes y pequeñas articulaciones, nefritis lúpica (síndrome nefrótico: proteinuria 3,28 gramos en 24 horas, albúmina sérica 1,2 g/dL), compromiso de piel y anexos (alopecia, lupus subagudo cutáneo), anticuerpos antinucleares (ANA) 1/1.250 patrón moteado y consumo de complemento C3 y C4. Dados los compromisos mencionados se consideró que cumplía con siete criterios (cinco clínicos y dos inmunológicos) clasificatorios para el diagnóstico de LES de acuerdo con SLICC (Systemic Lupus International Collaborating Clinics) al momento de la evaluación2.
En el año 2014 se documentaron síntomas secos (xerostomía y xeroftalmía), con queratoconjuntivitis sicca (confirmada por el servicio de oftalmología) asociada con la positividad para anti-SSa/Ro (147 UE/mL, vs. menor de 20 UE/mL) y anti-SSb/La (167 UE/mL, vs. menor de 20 UE/mL), realizándose el diagnóstico de SSs.
Presenta antecedentes asociados de hipertensión arterial y dislipidemia en tratamiento con losartán 50 mg cada 12 horas y atorvastatina 40 mg día, con manejo farmacológico específico para LES con cloroquina 250 mg día y prednisolona 10 mg día con inadecuada adherencia, con aparente control sintomático y clínico.
En mayo de 2016 ingresó al servicio de urgencias por presencia de tos con expectoración amarillenta de aproximadamente cuatro meses de evolución, pérdida de 10 kg de peso, diaforesis nocturna, deterioro de la clase funcional de 0/4 en escala de disnea modificada (mMRC) a 3/4, sin fiebre y con baciloscopias seriadas negativas. Al examen físico se evidenció: tensión arterial de 170/95, frecuencia cardiaca de 88 por minuto, frecuencia respiratoria de 18 por minuto, temperatura de 36,5°C, talla 145 cm, peso 47,1 kg, índice de masa corporal (IMC) de 22,5, saturación de O2 del 83%, la fracción inspirada de O2 de 0,21, la presencia de reforzamiento de S2 en foco pulmonar, estertores tipo velcro basales de predominio izquierdo, artritis de manos con limitación de arcos de movimiento.
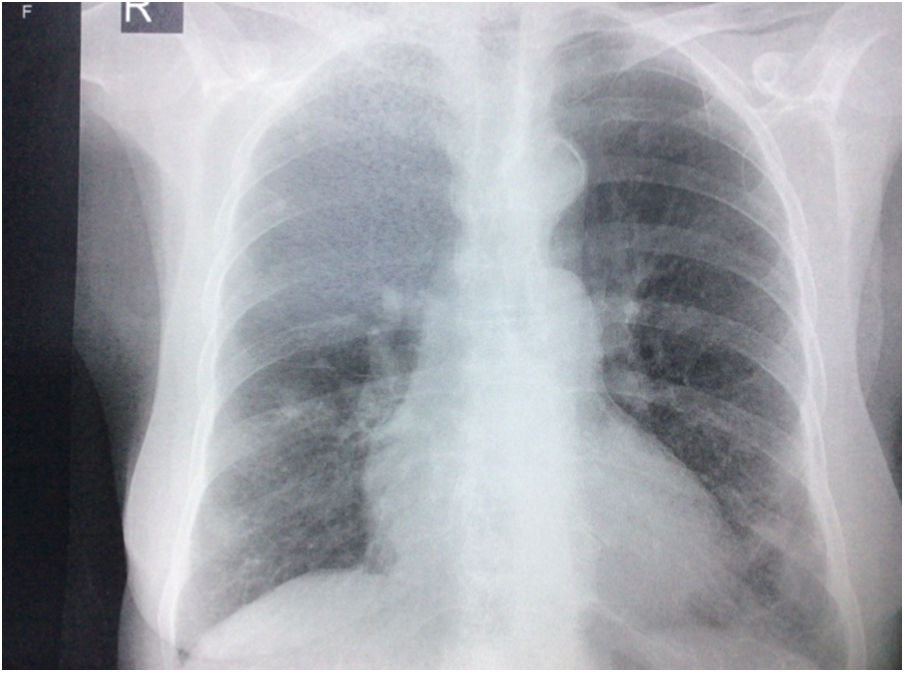
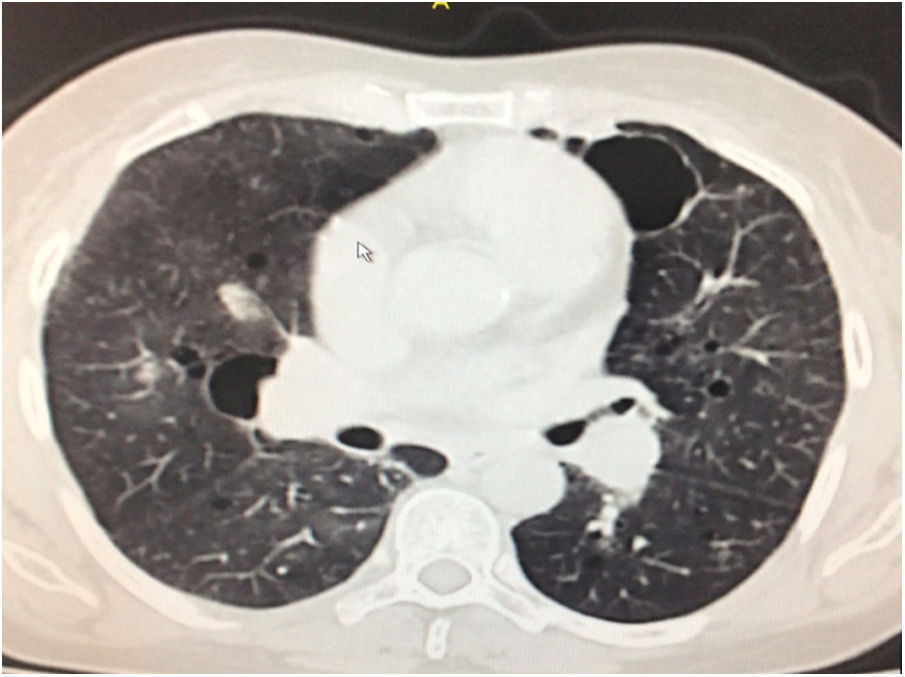
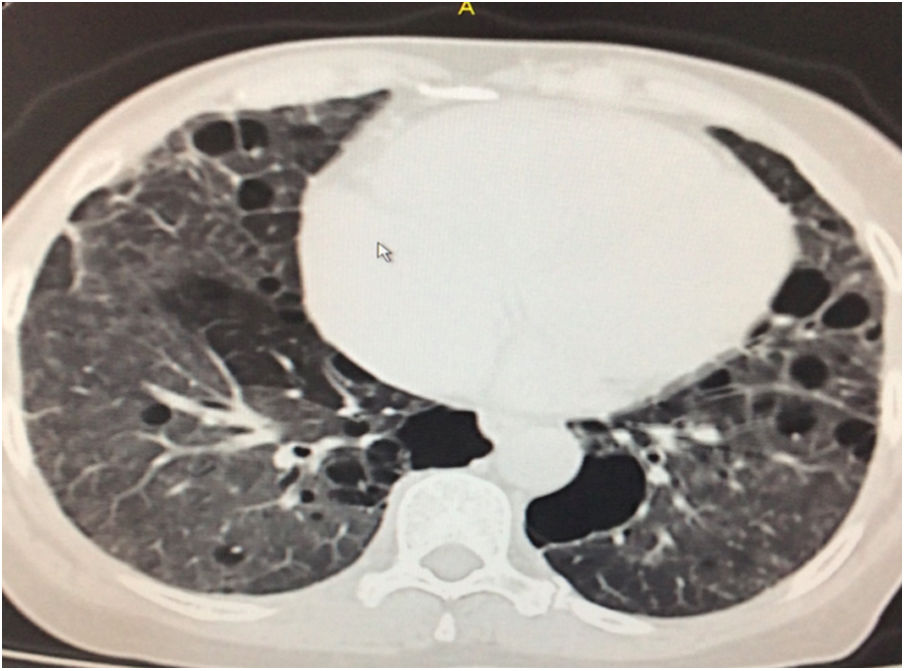
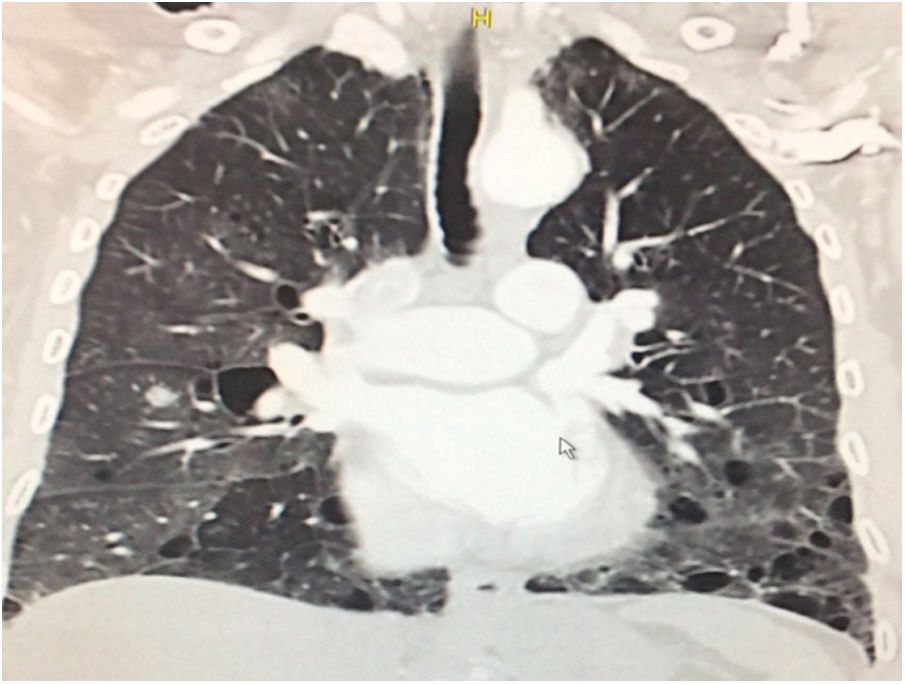
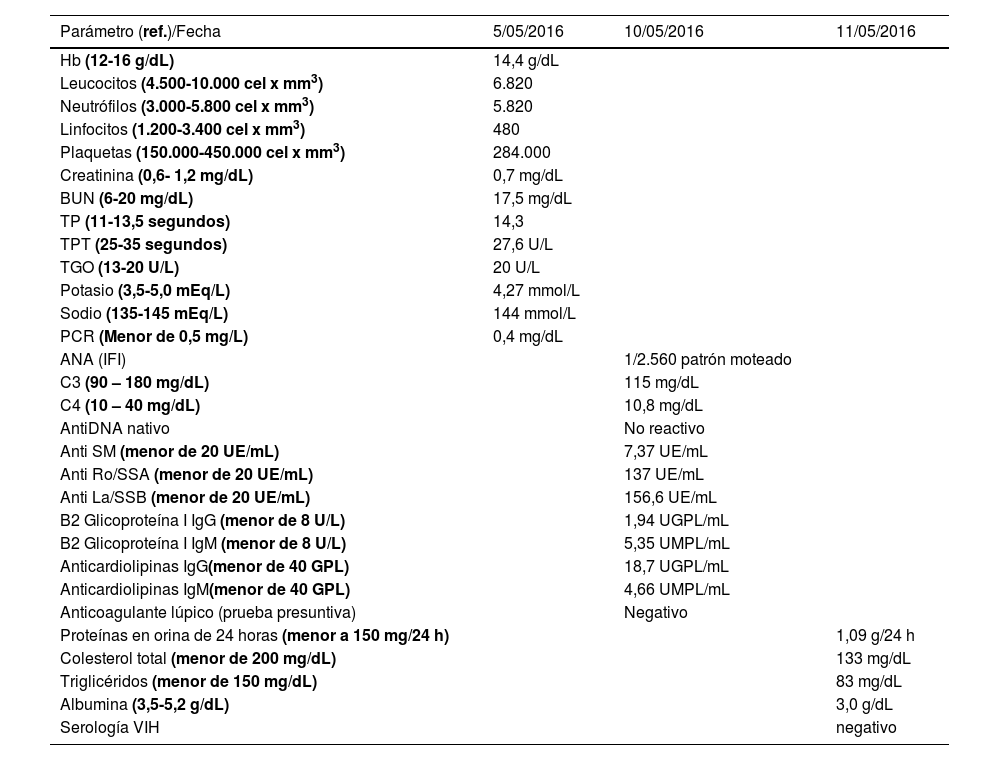
Durante la estancia hospitalaria los exámenes de laboratorio y las imágenes permitieron evidenciar (tabla 1): linfopenia con leucocitos normales, ANA positivos a título alto, complemento no consumido, proteinuria en rango subnefrótico, radiografía de tórax con presencia de opacidades de patrón intersticial de predominio en los 2/3 inferiores, micronodulares bilaterales y sin consolidaciones (fig. 1). Se realizó tomografía de tórax de alta resolución (TACAR) en la que se observaron múltiples quistes de pared delgada en ambos campos pulmonares, escasas zonas de atenuación en vidrio esmerilado subpleurales, engrosamiento septal y nódulos de distribución centrolobulillar (figs. 2-4). Se realizó un ecocardiograma transtorácico que reportó una fracción de eyección del ventrículo izquierdo del 70%, disfunción diastólica tipo I, hipertensión arterial pulmonar (PsAP) de 55 mmHg y desplazamiento sistólico del anillo tricuspídeo (siglas en inglés, TAPSE) de 2,0 cm (valor normal mayor de 1,6 cm).
Datos de laboratorio de la paciente durante la hospitalización
| Parámetro (ref.)/Fecha | 5/05/2016 | 10/05/2016 | 11/05/2016 |
|---|---|---|---|
| Hb (12-16 g/dL) | 14,4 g/dL | ||
| Leucocitos (4.500-10.000 cel x mm3) | 6.820 | ||
| Neutrófilos (3.000-5.800 cel x mm3) | 5.820 | ||
| Linfocitos (1.200-3.400 cel x mm3) | 480 | ||
| Plaquetas (150.000-450.000 cel x mm3) | 284.000 | ||
| Creatinina (0,6- 1,2 mg/dL) | 0,7 mg/dL | ||
| BUN (6-20 mg/dL) | 17,5 mg/dL | ||
| TP (11-13,5 segundos) | 14,3 | ||
| TPT (25-35 segundos) | 27,6 U/L | ||
| TGO (13-20 U/L) | 20 U/L | ||
| Potasio (3,5-5,0 mEq/L) | 4,27 mmol/L | ||
| Sodio (135-145 mEq/L) | 144 mmol/L | ||
| PCR (Menor de 0,5 mg/L) | 0,4 mg/dL | ||
| ANA (IFI) | 1/2.560 patrón moteado | ||
| C3 (90 – 180 mg/dL) | 115 mg/dL | ||
| C4 (10 – 40 mg/dL) | 10,8 mg/dL | ||
| AntiDNA nativo | No reactivo | ||
| Anti SM (menor de 20 UE/mL) | 7,37 UE/mL | ||
| Anti Ro/SSA (menor de 20 UE/mL) | 137 UE/mL | ||
| Anti La/SSB (menor de 20 UE/mL) | 156,6 UE/mL | ||
| B2 Glicoproteína I IgG (menor de 8 U/L) | 1,94 UGPL/mL | ||
| B2 Glicoproteína I IgM (menor de 8 U/L) | 5,35 UMPL/mL | ||
| Anticardiolipinas IgG(menor de 40 GPL) | 18,7 UGPL/mL | ||
| Anticardiolipinas IgM(menor de 40 GPL) | 4,66 UMPL/mL | ||
| Anticoagulante lúpico (prueba presuntiva) | Negativo | ||
| Proteínas en orina de 24 horas (menor a 150 mg/24 h) | 1,09 g/24 h | ||
| Colesterol total (menor de 200 mg/dL) | 133 mg/dL | ||
| Triglicéridos (menor de 150 mg/dL) | 83 mg/dL | ||
| Albumina (3,5-5,2 g/dL) | 3,0 g/dL | ||
| Serología VIH | negativo |

Radiografía de tórax con proyección postero-anterior en la que se observan opacidades intersticiales, presencia de nódulos con densidad cálcica de aproximadamente 8 a 9 mm en el tercio superior de hemitórax derecho, prominencia del botón y calcificación ateromatosa aórticas, además de cardiomegalia, sin derrames pleurales.
 que evidencia múltiples quistes de pared delgada de predominio central, zonas de atenuación en vidrio esmerilado subpleural y un nódulo calcificado de aproximadamente 6 mm de diámetro.")


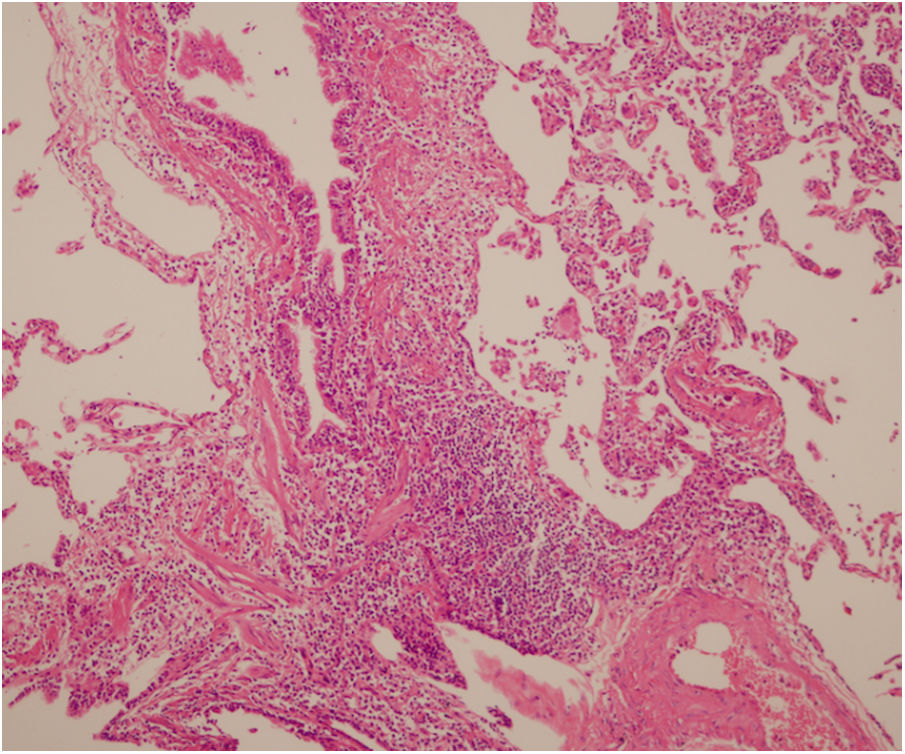
Se interpreta el patrón tomográfico como de una neumonía intersticial linfoide o linfocítica (NIL), se realiza biopsia transpulmonar que informó la presencia de infiltrado mononuclear de predominio linfocítico con histiocitos, que genera engrosamiento septal (fig. 5), compatible histológicamente con el diagnóstico de una NIL. La espirometría reportó una capacidad vital forzada (CVF) del 84%, un volumen espiratorio forzado en el primer segundo (VEF1) del 91% y una relación CVF/VEF1 del 91%. Con el diagnóstico radiológico e histológico de NIL y la presencia de proteinuria de 1 g en 24 horas, se decidió iniciar ciclofosfamida a dosis de 500 mg cada 15 días hasta completar seis dosis y un esquema de prednisolona oral de 50 mg al día. La paciente evolucionó adecuadamente durante la estancia hospitalaria, con mejoría de los síntomas respiratorios y sistémicos, excepto por la presencia de artritis persistente, por lo que se decidió adicionar metotrexate al tratamiento de base, dándosele egreso hospitalario al décimo día de estancia. Durante el seguimiento clínico ambulatorio se reduce la dosis de prednisolona a partir de la cuarta semana de tratamiento hasta llegar a una dosis de mantenimiento de 10 mg. Al sexto mes de tratamiento se documentó resolución completa de síntomas respiratorios, con clase funcional según mMRC de 0/4 y mejoría progresiva de la artritis, pero sin resolución completa de la misma, por lo que se ajustó la dosis de metotrexate.
Discusión
El compromiso pulmonar en el LES se consideró un evento terminal hasta la primera mitad del siglo xx. Sin embargo, durante la segunda mitad del siglo, se documentó en series anatomorradiológicas pulmonares de pacientes con LES que el compromiso pulmonar no era únicamente pleural ni terminal, como dictaba el concepto predominante de la época, sino todo lo contrario, también se podía hallar compromiso parenquimatoso en fases tempranas de la enfermedad3. En la década de los setenta del siglo xx aparecieron reportes apuntando a que la alteración pulmonar no era tan inusual, con una frecuencia de 11 a 15% en los pacientes con LES, incluso con descripciones de casos de compromiso pulmonar como presentación inicial de la enfermedad4,5. En 1979, Hunninghake y Fauci propusieron una clasificación que incluyó la pleuresía (derrame pleural de carácter inflamatorio), las atelectasias, el edema pulmonar urémico debido a deterioro de la función renal, la neumonitis lúpica aguda, la enfermedad intersticial difusa o enfermedad pulmonar intersticial (EPI) y la disfunción diafragmática con pérdida de volumen pulmonar (síndrome de pulmón encogido)6.
En el LES la EPI se ha documentado con una frecuencia del 6 al 11% de acuerdo con series clinicorradiológicas y de necropsias, con un franco compromiso fibrótico por histología hasta del 4% de los casos5,7. Se presenta con desarrollo progresivo de disnea y tos ligera con o sin expectoración, en un periodo de semanas a meses. La mayoría los pacientes con EPI asociada con el LES cursan con compromiso de otros sistemas, predominantemente osteoarticular, cutáneo, hematológico, renal, vasculitis y fenómeno de Raynaud8. El patrón histológico más frecuentemente reportado en la literatura de EPI, en enfermedades reumatológicas, es de neumonía intersticial no específica, tanto en LES como en Sd de Sjögren9,10, al igual que en otras patologías reumatológicas como las miositis inflamatorias y la esclerosis sistémica, entre otras.
La EPI asociada con el Sd de Sjögren secundario al LES (LES-SSs) no es infrecuente, informándose entre un 15 a 30%11,12 no muy diferente a lo reportado en LES sin presencia de SSs, de aproximadamente un 15%11. El patrón de NIL es ocasional y poco frecuente en el LES13,14 y en el Sd de Sjögren primario constituye aproximadamente del 8 al 16% de los casos reportados por histología en algunas series15,16. Sin embargo, Deheinzelin et al., informaron hasta el 42% de este tipo de patrón histológico en Sd de Sjögren primario17. La NIL fue descrita inicialmente como un seudolinfoma con una afección extraglandular en los pacientes con Sd de Sjögren18, es una entidad descrita también en pacientes con infección por VIH, hepatitis activa y colangitis (cirrosis) biliar primaria que histológicamente se caracteriza por la infiltración intersticial de linfocitos T policlonales, células plasmáticas, histiocitos, con expansión difusa de los septos alveolares e interlobulares e hiperplasia tipo MALT (de las siglas en ingles Mucossal-Asociated Lymphoid Tissue), con edades de presentación entre los 40 y 70 años de edad y un pico en la sexta década de la vida19,20. La imagen tomográfica de alta resolución habitualmente evidencia la presencia de áreas de consolidación en parches, nódulos centrolobulillares, atenuación en vidrio esmerilado e imágenes quísticas multifocales de predominio central15,21. La confirmación de NIL basada tanto por el patrón tomográfico como por la histología es fundamental dado el relativo buen pronóstico de esta entidad y su adecuada respuesta terapéutica15,22,23, siendo la microscopía óptica central en la evaluación diagnóstica, teniendo en cuenta la existencia de patologías con presentación imagenológica y patrones histológicos similares como el linfoma extranodal o la enfermedad de Castelman15,22,24-26, además del riesgo de evolución a linfoma de la NIL asociada al Sd de Sjögren. A este respecto, la literatura es heterogénea en definir la asociación de NIL y el desarrollo de linfoma a través del tiempo27-30. Algunos autores han sugerido que muy posiblemente los casos reportados de NIL que progresan a linfoma fueron realmente casos de linfoma inadecuadamente identificados en el trabajo diagnóstico inicial31.
La paciente en el presente caso se presenta con un cuadro de una EPI de evolución crónica asociada a un importante compromiso sistémico, artropatía de Jaccoud con artritis activa y nefritis con proteinuria de 1 g en 24 horas; similar a los reportes de series de casos con extenso compromiso sistémico asociado8,12, así como a positividad para anti-Ro/SSA y anti-La/SSB como una característica inmunológica relacionada al compromiso pulmonar intersticial del LES-SSs11. El tratamiento de esta asociación aún no es claro, la mayoría de la evidencia está soportada en reportes de casos con escaso número de pacientes y con una respuesta terapéutica en términos clínicos, sintomáticos o de los parámetros de la función pulmonar. Dichas terapéuticas están basadas primordialmente en el uso de esteroides y en los casos refractarios con el uso de ciclofosfamida o azatioprina8,15. Otros casos se han informado con el inicio de ciclofosfamida de forma temprana en los pacientes con otras indicaciones terapéuticas como la presencia de nefritis32.
Nuestra paciente evolucionó satisfactoriamente con el esquema farmacológico de corticosteroides y ciclofosfamida en pulsos mensuales, que llevó a la resolución de los síntomas respiratorios y restablecimiento de la clase funcional basal; sin embargo, con persistencia del compromiso inflamatorio articular y, ante la consideración riesgo-beneficio, se le inició metotrexate con una adecuada respuesta articular y sin evidencia de progresión del compromiso pulmonar.
FinanciaciónNinguna.
Conflicto de interesesLoa autores declaran no tener ningún conflicto de intereses.
