




El síndrome de Sjögren es una entidad multisistémica de naturaleza autoinmune, clásicamente considerada una exocrinopatía debido a la alta frecuencia de síntomas secos (queratoconjuntivitis seca, xerostomía) como resultado de infiltración poliglandular por linfocitos autorreactivos. Sin embargo, menos del 10% de estos pacientes puede iniciar con manifestaciones extraglandulares severas, traducidas en peores desenlaces a largo plazo. Se presenta el caso de una gestante que inició con síndrome de debilidad aguda proximal relacionada con miositis con enfermedad mitocondrial e hipopotasemia severa, en el contexto de acidosis tubular renal distal, como manifestación extraglandular de síndrome de Sjögren primario. Se discuten brevemente manifestaciones neurológicas de esta entidad, incluyendo aquellas secundarias a trastornos metabólicos precipitados por compromiso autoinmune.
Sjögren's syndrome is a multisystemic autoimmune disorder. It is classically considered as an exocrine disease, given the high frequency of dry symptoms (keratoconjunctivitis sicca, xerostomia) as a result of poly-glandular infiltration by autoreactive lymphocytes. However, less than 10% of these patients can onset with severe extra-glandular manifestations, resulting in worse long-term outcomes. The case of a pregnant woman is presented, who debuted with acute proximal weakness syndrome related to myositis with mitochondrial pathology and severe hypokalaemia in the context of distal renal tubular acidosis, as an extra-glandular manifestation of primary Sjögren's syndrome. Neurological manifestations of this condition are briefly discussed, including those secondary to metabolic disorders precipitated by autoimmune compromise.
El síndrome de Sjögren primario (SSp) es una enfermedad autoinmune inflamatoria crónica, caracterizada por infiltración linfocitaria de glándulas exocrinas, en asociación con la producción de anticuerpos antinucleares (ANA) como anti-Ro y anti-La1,2, lo que da como resultado disfunción glandular y síntomas secos3.
La prevalencia global del SSp puede llegar a ser de hasta del 4,6% en algunas series; sin embargo, esto depende de los criterios diagnósticos usados en las diferentes áreas geográficas. En Europa, la prevalencia varía de manera significativa, con cifras del 2,7% en Suiza, el 0,03% y el 0,01% en Italia y Francia, respectivamente4,5.
En Latinoamérica, donde los reportes son heterogéneos, se han encontrado prevalencias que van desde el 0,17% en Brasil hasta el 0,02% en Guatemala. En Colombia, son pocos los estudios de prevalencia en enfermedades reumatológicas; se cuenta con datos de 2estudios epidemiológicos recientes que muestran una prevalencia general del SSp entre el 0,08 y el 0,12% en el país4,6.
De la totalidad de estos pacientes, alrededor del 30 al 40% presentará hallazgos sistémicos extraglandulares, de los cuales las manifestaciones neurológicas corresponden a entre el 10 y el 40%. Entre el 10 y el 30% de los pacientes iniciarán con una presentación neurológica como manifestación inicial del SSp2,5.
Aproximadamente el 80% de los pacientes diagnosticados de SSp se presenta con algún tipo de síntoma seco, siendo el más común ojo seco, seguido de boca seca1; de estos, del 30 al 40% presenta manifestaciones sistémicas extraglandulares. Las manifestaciones neurológicas varían en frecuencia; sin embargo, el inicio del SSp con alguna de estas tiene baja prevalencia2. De forma ocasional, los pacientes con SSp se presentan con debilidad y mialgias, escenario en el cual se debe considerar una miopatía inflamatoria, ya sea secundaria al SSp, o como una manifestación secundaria a otra enfermedad reumatológica, enmarcado dentro de un síndrome de poliautoinmunidad2.
Presentamos el caso de una gestante de 40 años, sin antecedentes patológicos conocidos, que inició con debilidad aguda de predominio proximal y se encontró hipopotasemia severa asociada y acidosis tubular renal distal. Tras ello se ampliaron los estudios, los cuales cumplían criterios ACR-EULAR 2017 para SSp5. Dada la elevación de la CK total en contexto de mialgias previas al cuadro de debilidad aguda, se realizó biopsia muscular, la cual mostró cambios miopáticos con anomalías mitocondriales. Luego de este hallazgo, se hizo una descripción de las manifestaciones neurológicas del síndrome, con énfasis en los hallazgos miopáticos asociados al SSp.
Se diligenció consentimiento informado por parte de la paciente.
Caso clínicoMujer de 40 años, natural y procedente de Leticia, Amazonas, sin antecedentes patológicos conocidos, con embarazo de semanas, quien 2meses antes del ingreso presentó cuadro de debilidad proximal de las 4extremidades, que la obligó a permanecer en cama durante días, con posterior recuperación espontánea. A los 10 días presentó nuevamente debilidad, de inicio en miembros inferiores, que luego comprometía miembros superiores y se asociaba con mialgias intensas. Tras ello presentó disfagia y en pocos días falla ventilatoria que requirió intubación orotraqueal y remisión a nuestra institución.
Al ingreso se encontraba intubada, con ventilación mecánica invasiva, febril, taquicárdica, normotensa (presión arterial: 100/54mmHg), con edemas y dolor a la palpación muscular. En el examen neurológico se mostró somnolienta por sedación con dexmedetomidina, fuerza flexoextensión de cuello en 1/5, miembros superiores proximal 0/5, distal 2/5 y miembros inferiores en 1/5, arreflexia generalizada, respuesta plantar neutra bilateral, sin alteraciones sensitivas. Se consideró, inicialmente, una polineuropatía aguda, teniendo en cuenta la arreflexia; sin embargo, se consideró una miopatía como causa de la debilidad, debido al compromiso motor puro y las mialgias. La paciente fue valorada por ginecobstetricia tras lo cual se documentó embarazo de 10,5 semanas por ecografía de primer trimestre.
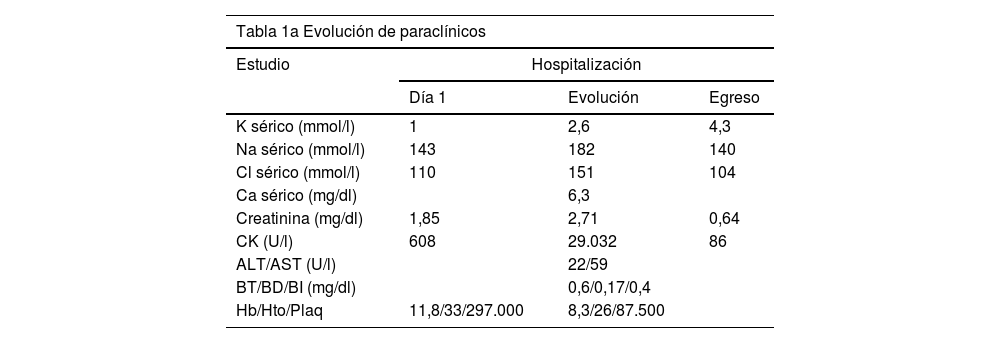
Paraclínicos de sitio de remisión con leucocitosis neutrofilica, hipopotasemia severa y magnesio sérico en límite superior de normalidad, además de azoados elevados, CK en 608 U/l y gases arteriales con acidosis metabólica compensada, En vista de la hipopotasemia severa asociada a acidosis metabólica hiperclorémica, con anión GAP normal, se consideró probable acidosis tubular renal distal (tipo i), aunque no se pudo calcular el gradiente transtubular de potasio de manera confiable pues en ese momento la paciente ya se encontraba en reposición de este electrólito por catéter venoso central y no se contaba con cálculo de sodio urinario ni osmolaridad urinaria (tabla 1a y c).
Resultados de laboratorio
| Tabla 1a Evolución de paraclínicos | |||
|---|---|---|---|
| Estudio | Hospitalización | ||
| Día 1 | Evolución | Egreso | |
| K sérico (mmol/l) | 1 | 2,6 | 4,3 |
| Na sérico (mmol/l) | 143 | 182 | 140 |
| Cl sérico (mmol/l) | 110 | 151 | 104 |
| Ca sérico (mg/dl) | 6,3 | ||
| Creatinina (mg/dl) | 1,85 | 2,71 | 0,64 |
| CK (U/l) | 608 | 29.032 | 86 |
| ALT/AST (U/l) | 22/59 | ||
| BT/BD/BI (mg/dl) | 0,6/0,17/0,4 | ||
| Hb/Hto/Plaq | 11,8/33/297.000 | 8,3/26/87.500 | |
| Tabla 1b Perfil inmunológico | |
|---|---|
| ANA | 1/2.560 moteado |
| Anti-Ro SSA (U) | 127,9, positivo fuerte |
| Anti-La SSB (U) | Negativo |
| Anti-Sm (U) | Negativo |
| Anti-RNP (U) | Negativo |
| C3/C4 | 80,1/26,8 |
| Ac. anti-PR3 | 1,7 |
| Ac. anti-MPO | 1,3 |
| Electroforesis de proteínas | Hipergammaglobulinemia policlonal, hipoalbuminemiaAlbúmina sérica: 2 g/dl |
| Tabla 1c Química sanguínea | |
|---|---|
| K orina aislada (mmol/l) | 21,2 |
| Na orina aislada (mmol/l) | 59,3 |
| Cl orina aislada (mmol/l) | 74,7 |
| Ca orina aislada (mg/dl) | < 1 |
| Creatinuria aislada (mg/dl) | 92 |
| BUN orina aislada (mg/dl) | 465 |
Desde el punto de vista clínico, la evolución fue estacionaria, con picos febriles intermitentes, sin evidencia de foco infeccioso claro, incluyendo punción lumbar que descartó infección en el sistema nervioso central (SNC). La paciente presentó elevación progresiva de la CK hasta 29.000 UI/l, acidosis metabólica severa en compensación con pH de 7,1, PCO2: 70 y HCO3: 22,2 con BE de –7,3, y a la semana del ingreso presentó aborto espontáneo; se descartó etiología séptica relacionada.
Posteriormente, cursó con deterioro multisistémico, compromiso renal multifactorial y falla ventilatoria que requirió bomba de circulación de membrana extracorpórea (ECMO) veno-venosa y terapia de reemplazo renal. Después de 7 días se logró el desmonte de la ECMO y se pasó a tienda de traqueostomía, con persistencia de cuadriparesia flácida arrefléxica, a pesar de adecuada reposición electrolítica. Por compromiso deglutorio persistente, requirió gastrostomía.
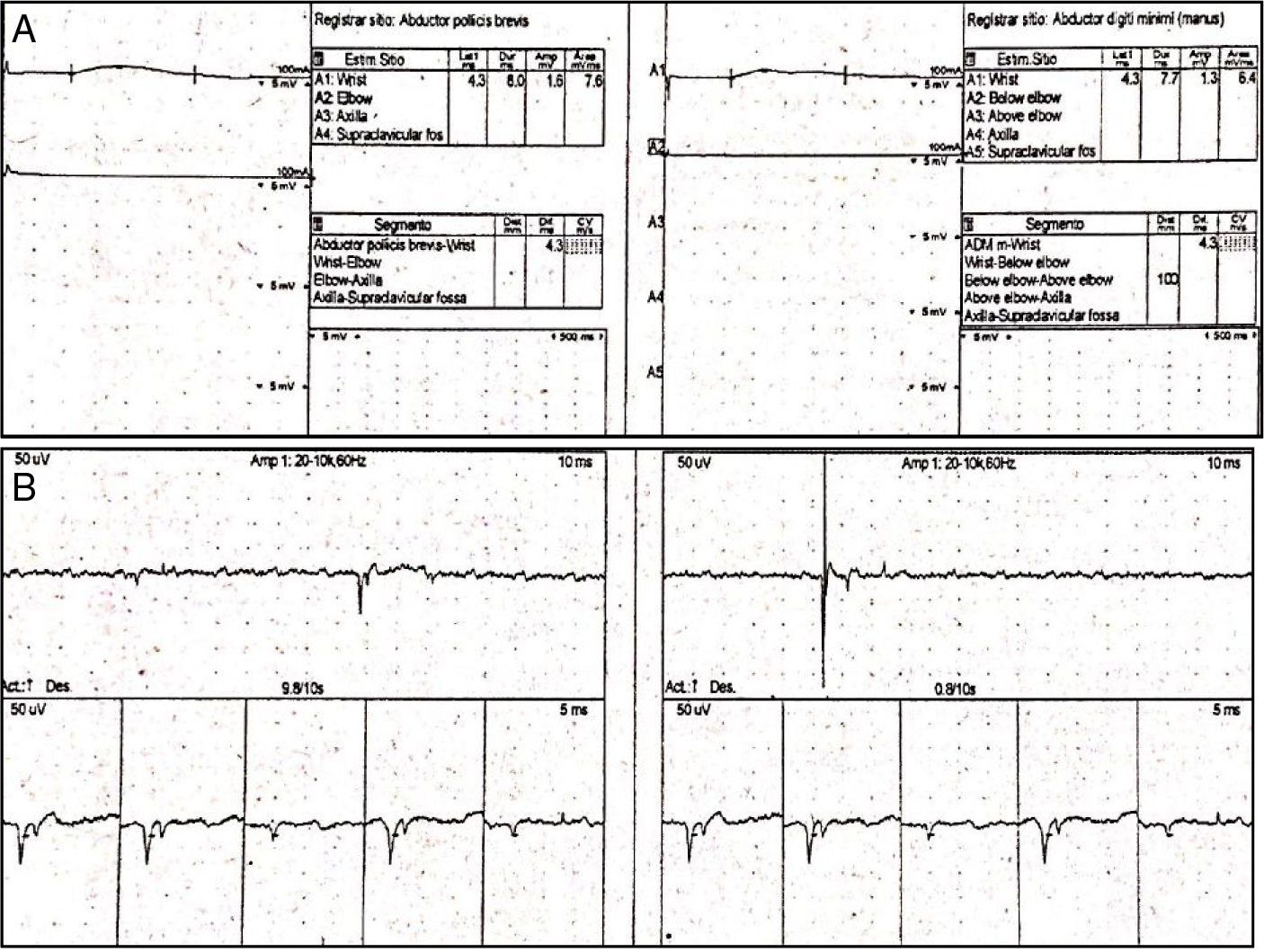
Las neuroconducciones mostraron una polineuropatía sensitivo-motora axonal. La electromiografía fue técnicamente difícil de realizar dada la imposibilidad de realización de fuerza máxima por parte de la paciente y también por su estado de conciencia; sin embargo, se observó un patrón miopático adicional al compromiso neuropático documentado por neuroconducción (fig. 1A y B). Adicionalmente, con ANA positivos en 1/2.560 (patrón moteado) y anti-Ro en 127,9, fuertemente positivo, orientando hacia una etiología autoinmune (tabla 1b), por lo cual se realizó biopsia de glándula salivar menor que mostró sialoadenitis linfocítica focal y puntaje focal de > 1 foco/4mm2 y un test de Schirmer positivo para ojo seco bilateral.
 Neuroconducciones. Izquierda: nervio mediano izquierdo. Derecha: nervio cubital izquierdo. Se observa degeneración axonal sensitivo-motora para los nervios medianos, cubital, tibial, peroneo y sural. Estos hallazgos son simétricos e indican una polineuropatía sensitivo-motora. B) Electromiografía. Se observan ondas agudas positivas y fibrilaciones, compatibles con un patrón miopático con potenciales pequeños, reclutamiento temprano e inestabilidad de membrana.")
A) Neuroconducciones. Izquierda: nervio mediano izquierdo. Derecha: nervio cubital izquierdo. Se observa degeneración axonal sensitivo-motora para los nervios medianos, cubital, tibial, peroneo y sural. Estos hallazgos son simétricos e indican una polineuropatía sensitivo-motora. B) Electromiografía. Se observan ondas agudas positivas y fibrilaciones, compatibles con un patrón miopático con potenciales pequeños, reclutamiento temprano e inestabilidad de membrana.
Debido a la persistencia de la debilidad, así como a la elevación exponencial de los niveles de CK total, se realizó una biopsia muscular de cuádriceps izquierdo que reportó cambios miopáticos con anomalías mitocondriales y acúmulo de lípidos, presencia de fibras rojas rasgadas y expresión de HLA-I en algunas fibras musculares. Por tal motivo, se consideraron dentro del diagnóstico diferencial desórdenes del metabolismo de lípidos, así como miopatías asociadas a enfermedad autoinmune como la polimiositis con enfermedad mitocondrial (PM-Mito) y la miositis por cuerpos de inclusión (MCI). Fue valorada por genética clínica, que llevó a cabo perfil de acil-carnitinas y lactato/piruvato, que estaban dentro de límites normales. Los antecedentes familiares ante sospecha de miopatía metabólica fueron negativos.
Por enfermedad y biomarcadores, se consideró que la paciente cumplía criterios para diagnóstico de SSp (tabla 2), con manifestaciones extraglandulares neurológicas, tanto de orden muscular como de nervio periférico. Se excluyeron precipitantes tóxicos o farmacológicos (medicamentos con efecto anticolinérgico durante el curso agudo de la enfermedad). En este contexto, se inició manejo con prednisolona 1mg/kg/día y azatioprina 50mg/día. En el tránsito del proceso de rehabilitación física se lograron retirar la traqueostomía y la gastrostomía, y se observó tolerancia de la vía oral mediante terapia deglutoria intensiva, así como una mejoría progresiva de la fuerza de las extremidades, hasta lograr fuerza 3/5 proximal y conseguir la deambulación asistida. Después de 5meses de hospitalización, la paciente fue remitida a un centro de menor nivel de complejidad en su ciudad de origen, para continuar la rehabilitación integral.
Criterios diagnósticos para SSp (ACR-EULAR 2017a) aplicados al caso clínico
| Ítem | Descripción | Puntaje |
|---|---|---|
| Focus score ≥ 1 | Una puntuación determinada por el número de infiltrados de células mononucleares que contiene ≥ 50 células inflamatorias por 4mm2 de menor glándula salival labial obtenida en biopsia | 3b |
| Anticuerpos anti-SSAc | Medido en suero; solo se deben considerar los anticuerpos anti-Ro60; los anticuerpos anti-Ro52 aislados no son específicos para SSp | 3 |
| Puntaje de tinción ocular SICCA ≥ 5 | Puntuación determinada por un oftalmólogo con base en el examen con tinción con fluoresceína y lisamina verde; los puntajes varían de 0 a 12, con puntajes más altos que indican mayor severidad | N/A |
| Test de Schirmer | Ensayo para medir la producción de lágrimas mediante la inserción de papel de filtro en conjuntiva del párpado inferior; evalúa cantidad de humedad en el papel | 1 |
| Flujo salival completo no estimulado de ≤ 0,1ml por min | Ensayo para medir la velocidad del flujo salival mediante la recolección de saliva en un tubo durante al menos 5min después de que el paciente haya deglutido | N/A |
| Puntaje total | 7/9 |
ACR: American College of Rheumatology; EULAR: European League Against Rheumatism; SICCA: Sjögren's International Collaborative Clinical Alliance; SSA: anti Sjögren's syndrome antígeno relacionado A.
Sobre la base de los criterios de clasificación enumerados, un diagnóstico de síndrome de Sjögren primario se define como una puntuación de 4 o más. Estos criterios se aplican a pacientes que tienen al menos un síntoma de sequedad ocular u oral o la presencia manifestaciones indicativas de síndrome de Sjögren primario. Los criterios de exclusión incluyen infección activa por el virus de la hepatitis C en el ensayo de reacción en cadena de la polimerasa, radioterapia de la columna cervical, sarcoidosis, enfermedad de injerto contra huésped, uso de fármacos anticolinérgicos y enfermedad relacionada con IgG4.
Los resultados serológicos positivos para los anticuerpos anti-SSA, SSB/La en ausencia de anticuerpos anti-SSA/Ro no son específicos y ya no se consideran criterio diagnóstico.
Modificado de Mariette y Criswell5.
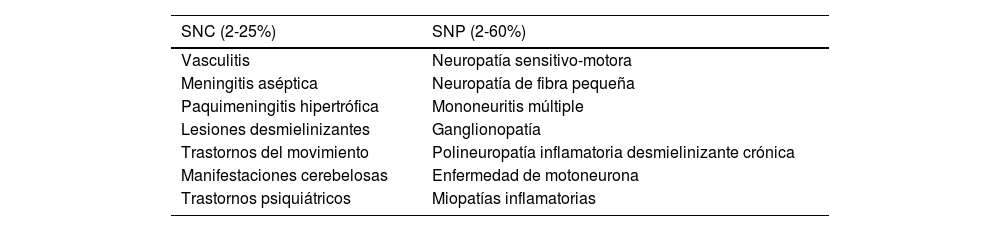
Las múltiples manifestaciones neurológicas del SSp comprometen tanto el SNC como las raíces nerviosas, el nervio periférico o el músculo. Dentro de los diferentes hallazgos neurológicos, el más frecuente es la neuropatía periférica, con frecuencia reportada del 2 al 60%7,8. La neuropatía sensitiva mixta (sensitiva y motora), la neuropatía craneal y la radiculopatía son algunas de las diferentes afectaciones del sistema nervioso periférico. Sin embargo, la neuropatía más frecuente es la sensitiva pura desmielinizante2,7 (tabla 3).
Manifestaciones neurológicas del síndrome de Sjögren primario
| SNC (2-25%) | SNP (2-60%) |
|---|---|
| Vasculitis | Neuropatía sensitivo-motora |
| Meningitis aséptica | Neuropatía de fibra pequeña |
| Paquimeningitis hipertrófica | Mononeuritis múltiple |
| Lesiones desmielinizantes | Ganglionopatía |
| Trastornos del movimiento | Polineuropatía inflamatoria desmielinizante crónica |
| Manifestaciones cerebelosas | Enfermedad de motoneurona |
| Trastornos psiquiátricos | Miopatías inflamatorias |
Las manifestaciones en el SNC son menos prevalentes, con frecuencias que van del 2 al 25%; sin embargo, en algunas series alcanzan hasta el 70%9. Clínicamente, es un amplio espectro de signos y síntomas que van desde un trastorno cognitivo, meningitis aséptica, lesiones inflamatorias desmielinizantes de predominio en fosa posterior, encefalitis subaguda, neuritis óptica y lesiones medulares, hasta movimientos anormales por lesiones gangliobasales2,7,10.
El síndrome de debilidad aguda y subaguda como presentación neurológica del SSp es un hallazgo poco frecuente pero altamente relevante, tanto por el diagnóstico diferencial como por las implicaciones clínicas asociadas. Algunos pacientes se pueden presentar con mialgias, fatiga y debilidad de predominio proximal, por lo que se deben considerar miopatías inflamatorias, entre las cuales la MCI es la más frecuentemente asociada a SSp, ya sea secundaria o como expresión de un síndrome de poliautoinmunidad con otra enfermedad subyacente2,7,11.
De igual manera, otros pacientes pueden presentarse con cuadriparesia, hipotonía, hipo o arreflexia, sin mialgias asociadas, caso en el cual es preciso considerar el espectro de neuropatías sensitivo-motoras o radiculopatías asociadas al SSp. Sin embargo, hay reportes de casos de pacientes que se presentaron con cuadriparesia flácida y se encontró hipopotasemia severa asociada a acidosis tubular renal distal, también como expresión de compromiso por el SSp12,13.
La MCI se presenta con debilidad muscular progresiva, tanto distal como proximal, que compromete principalmente los extensores de la rodilla y los flexores de los dedos. La PM-Mito comparte similitudes con diferentes grados de severidad en su presentación, las más de las veces con un patrón de debilidad proximal. Las 2entidades se presentan con debilidad focal y parchada que tiende a generalizarse con posterioridad11.
Desde el punto de vista clínico, la PM-Mito tiene un curso más lento de instauración de la debilidad progresiva que la MCI y rara vez tiene agregados de tinción TDP-43 o SMI-31 en fibras musculares. Las proteínas más frecuentes en los agregados tanto en PM-Mito como en MCI son LC3, un marcador de autofagia, y la cadena beta de α-cristalina, pequeña proteína de choque térmico14. Las alteraciones en las vías de degradación autofágica pueden ser un mecanismo patogénico común entre ambas entidades. En general, la tinción de actividad enzimática mitocondrial, agregados y las células B ayudan a distinguir PM-Mito de los síndromes de miopatía inflamatoria convencionales, que tienen más probabilidades de responder al tratamiento con esteroides15.
No hay reportes en la literatura local de pacientes con miopatía inflamatoria como causa de un cuadro de debilidad aguda en el inicio de un SSp, confirmada por patología. Las miopatías mitocondriales tipo PM-Mito, en el contexto de las enfermedades autoinmunes, han sido discutidas y revisadas en varios reportes de caso, sin contar aún con criterios diagnósticos establecidos para diferenciarlas de la MCI, ya que tienen varias similitudes tanto clínicas como histológicas.
Algunos autores han llegado incluso a postular la PM-Mito como una variante o una forma temprana de la MCI15, sin embargo, la presencia de depósitos de amiloide y vacuolas en anillo es más característica de la MCI que de la PM-Mito15. Los estudios de ADN mitocondrial muestran gran cantidad de deleciones, lo que refleja el componente mitocondrial tanto en la PM-Mito como en la MCI y pone en evidencia las similitudes en su mecanismo fisiopatogénico, pero sin especificidad para ninguna de las 2entidades15,16.
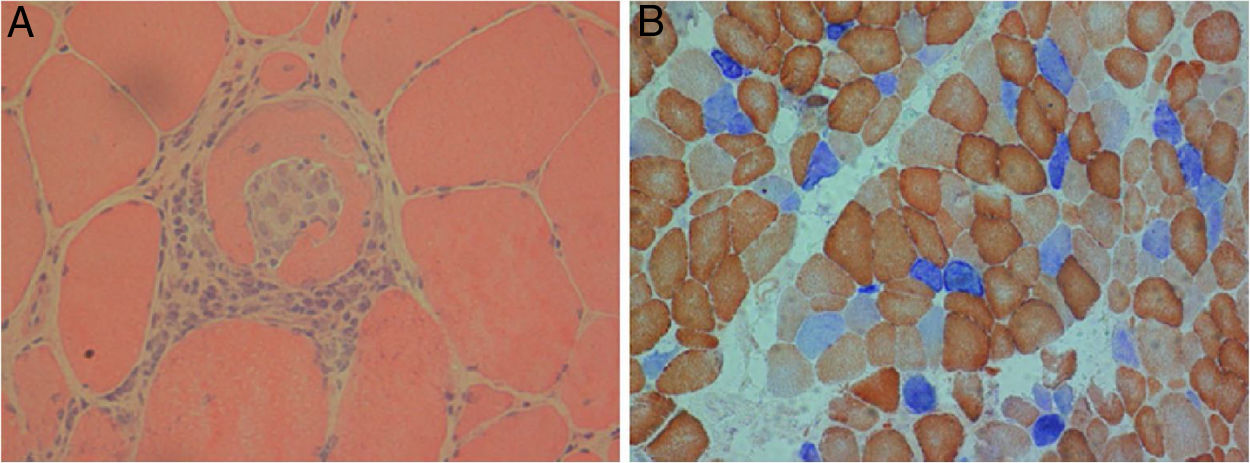
En la histopatología se evidencia inflamación mediada por células T, de predominio endomisial, así como expresión de moléculas HLA-I, moléculas coestimuladoras, vacuolas y agregados proteicos. La enfermedad mitocondrial se manifiesta histoquímicamente por dispersión de las fibras musculares con disminución de la citocromo-oxidasa, cuya deficiencia es una característica de esta entidad, más frecuente en la PM-Mito17 (fig. 2).
. Secciones de biopsia muscular teñidas con hematoxilina-eosina que muestran infiltrados endomisiales (a) y células inflamatorias invasivas, una fibra muscular no necrótica y una tinción COX-SDH (×40), que revela muchas fibras musculares con deficiencia de citocromo oxidasa (COX) (×20) (b). Tomada, con previa autorización, de Papadimas et al.22.")
Biopsia muscular de polimiositis con enfermedad mitocondrial (PM-Mito). Secciones de biopsia muscular teñidas con hematoxilina-eosina que muestran infiltrados endomisiales (a) y células inflamatorias invasivas, una fibra muscular no necrótica y una tinción COX-SDH (×40), que revela muchas fibras musculares con deficiencia de citocromo oxidasa (COX) (×20) (b).
Tomada, con previa autorización, de Papadimas et al.22.
En un amplio número de estudios se ha evaluado la relación entre autoanticuerpos séricos y las miopatías inflamatorias idiopáticas como la dermatomiositis y la polimiositis. Estas son enfermedades sistémicas de naturaleza autoinmune que se caracterizan por inflamación y disfunción del músculo esquelético, y lesiones en piel características de la dermatomiositis y se diferencian, principalmente, por hallazgos inmunohistopatológicos.
Dentro de las alteraciones serológicas, existen autoanticuerpos asociados (MAA por sus siglas en inglés) y específicos (MSA por sus siglas en inglés) de las miopatías inflamatorias, que son marcadores útiles para el diagnóstico y pronóstico. Dentro de los MSA se encuentran: anti-Jo, anti-Mi-2, anti-MDA5 y anti-SRP, entre otras, y los MAA corresponderían, en particular, a anti-Ro, anti-U1RNP, anti-PM/Scl y anti-Ku18-20.
Temmoku et al. describieron la prevalencia y el significado clínico de los MMA y MSA en un grupo de pacientes con miopatías inflamatorias y encontraron que la combinación de anti-MDA5 y anti-Ro era un marcador de mal pronóstico en dermatomiositis18. Así mismo, Ghirardello et al. caracterizaron perfiles séricos de autoanticuerpos de pacientes con miopatías inflamatorias y hallaron positividad del 50% para MSA y del 39% para MMA, de los cuales el 28% fue positivo para anti-Ro20.
No hay reportes de autoanticuerpos o perfiles séricos en miopatías no inflamatorias y la mayoría de los hallazgos de positividad de anti-Ro en miopatías habla del pronóstico de la enfermedad, ya que el objetivo de estos estudios no fue describir asociación con otras enfermedades autoinmunes, por lo cual su positividad no descarta la relación con el síndrome de Sjögren en presencia de miopatías como un síndrome de poliautoinmunidad18-21.
Papadimas et al. reportaron 4casos de mujeres con diagnóstico histopatológico de PM-Mito, que clínicamente cursaron con debilidad muscular proximal y simétrica. Se documentaron además deleciones a gran escala en el ADN mitocondrial. Una de estas pacientes tenía diagnóstico de SSp de 3años de evolución y desde entonces había iniciado con episodios autolimitados de debilidad muscular en miembros inferiores, al igual que en nuestro caso. En el examen neurológico se describía sutil caída cefálica, con debilidad muscular simétrica tanto proximal como distal leve en 4extremidades (flexión bilateral del codo, flexión de la muñeca, flexión de cadera 4/5, dorsiflexión derecha del pie 4/5, dorsiflexión izquierda del pie 4/5)22.
En el caso referenciado, el estudio electromiográfico reveló cambios miopáticos en la mayoría de los músculos examinados, con descargas espontáneas de potenciales de fibrilación y ondas agudas positivas, similar al registro de nuestra paciente (fig. 1 A y B). El valor máximo de CK total fue de 538 U/l, con velocidad de sedimentación globular en 45mm/h y panel de autoinmunidad con ANA positivos (1/320), sin que se describiera patrón y anticuerpos anti-La/SSB positivos, con anti-ADN, factor reumatoide y anti-Ro/SSA ausentes (a diferencia de nuestro caso). El panel para autoanticuerpos específicos en miositis inflamatorias fue negativo. Se inició manejo con esteroides (metilprednisolona, 64mg/día) + metotrexato 25mg/semana, con mejoría clínica gradual, hasta la resolución completa de los síntomas a los 4años del seguimiento22.
Lograr distinguir entre la MCI y la PM-Mito puede tener alguna utilidad pronóstica, principalmente porque la segunda tiene una progresión más lenta de la debilidad en meses o incluso años. La respuesta al tratamiento con esteroides e inmunomoduladores no es óptima en la mayoría de los reportes de caso, lo cual concuerda con el comportamiento de la MCI; sin embargo, al iniciar el tratamiento para la enfermedad autoinmune de base, en este caso, para el SSp, se puede encontrar mejoría progresiva de los signos y los síntomas de la enfermedad22-24.
ConclusiónEl SSp es una entidad autoinmune de curso crónico, que puede iniciar con compromiso extraglandular extenso, incluyendo el SNC y el sistema nervioso periférico, por lo que se debe considerar en pacientes con síndrome de debilidad aguda, de acuerdo con el contexto clínico específico, tras haber excluido las causas más frecuentes.
Desde el punto de vista clínico, en este caso fue difícil localizar topográficamente una única lesión dentro del sistema nervioso periférico; se observó un compromiso tanto neuropático como miopático. La paciente cursó con una polineuropatía sensitivo-motora aguda axonal, probablemente relacionada con el síndrome de Sjögren, pero además presentó una canalopatía adquirida, precipitada por una probable acidosis tubular renal distal (no confirmada) como causa de hipopotasemia severa y, adicionalmente, un compromiso muscular inflamatorio de manera simultánea.
De acuerdo con los hallazgos de la biopsia muscular y la presencia de autoinmunidad, se planteó que la miopatía pudo ser expresión de una enfermedad mitocondrial, ya fuera por la PM-Mito, o por una variante temprana de la MCI asociada al SSp. Se consideró el embarazo como un factor desencadenante de la respuesta inmunológica de la paciente. Así mismo, se consideró que el SSp es un diagnóstico de exclusión en un paciente críticamente enfermo.
Las manifestaciones extraglandulares del SSp son heterogéneas y no tienen una asociación lineal con los síntomas secos en el tiempo de evolución de la enfermedad. Desde el punto de vista neurológico, cuando no sea posible localizar topográficamente una única lesión en el sistema nervioso, se debe considerar este diagnóstico, ya que algunos pacientes con SSp presentan diferentes manifestaciones extraglandulares de forma simultánea (miopatía, canalopatía y neuropatía) que pueden retrasar su diagnóstico y manejo, con implicaciones pronósticas en el tiempo.
Consideramos que el espectro autoinmune propio de las miopatías inflamatorias es un área aún en desarrollo, que requiere equipos multidisciplinarios de investigación, con especial participación de las ciencias básicas, debido al daño mitocondrial transversal a estas enfermedades y el compromiso multiorgánico relacionado. Este caso destaca la importancia de reconocer el compromiso neuromuscular múltiple y simultáneo de entidades tan complejas como el SSp25-27.
FinanciaciónNinguna.
Conflicto de interesesLos autores no declaran conflicto de intereses.
Consentimiento informadoEl consentimiento informado fue debidamente diligenciado por parte de la paciente, con respeto por el derecho a la privacidad. Se recibió aval por parte del Comité de Ética del Hospital Universitario San Ignacio para su publicación en revista científica.
Agradecemos al Dr. Pablo Gil, internista neumólogo, y a la Dra. María del Mar Bautista, residente de Medicina Interna, por su colaboración en el seguimiento del caso clínico. Así mismo, al Hospital Universitario San Ignacio.
