





El síndrome de Sjögren primario (SSp) es una enfermedad autoinmune que afecta principalmente al tejido glandular. A pesar de ello, puede involucrar otros sistemas, siendo el compromiso neuropsiquiátrico una manifestación extraglandular común. Su presentación clínica varía ampliamente según el dominio que se encuentre afectado, y por tanto puede dividirse en tres grandes categorías: sistema nervioso central, sistema nervioso periférico y psiquiátrico. Algunas de estas complicaciones comparten mecanismos fisiopatológicos comunes, entre los principales la vasculitis/vasculopatía, la infiltración linfocítica y la presencia de anticuerpos antineuronales. La diversidad en la presentación clínica de esta entidad impide hacer una aproximación diagnóstica común, por lo cual la utilización de estudios específicos depende de un adecuado reconocimiento y de la localización por parte del clínico. El tratamiento debe dirigirse al mecanismo fisiopatológico implicado y, de acuerdo con el tipo de manifestación, puede incluso estar limitado al manejo sintomático.
Primary Sjögren's syndrome is an autoimmune disease that mainly involves glandular tissue. Despite this, it can potentially develop systemic involvement, within which neuropsychiatric manifestations are common. The clinical presentation may vary widely depending on the domain affected, and may thus be classified into three categories: central nervous system, peripheral nervous system, and psychiatric. Some of these complications share a common pathophysiology, amongst which are vasculitis/ vasculopathy, lymphocytic infiltration and positive antineuronal antibodies. The wide clinical presentation makes it difficult to establish a common diagnostic approach, making it essential for the clinician to recognise and localise the type of compromise, so that diagnostic tools can be more advantageously employed. Treatment must be directed towards the underlying pathophysiology, and depending on the type of compromise, it can even be limited solely to the management of symptoms.
El síndrome de Sjögren primario (SSp) es una enfermedad crónica sistémica de etiología autoinmune que suele manifestarse con xeroftalmia y xerostomía, lo cual se conoce clásicamente como «síntomas sicca». La presentación clínica puede variar desde formas locales «benignas» (afectación de glándulas exocrinas) hasta manifestaciones sistémicas o también llamadas extraglandulares (pulmonares, gastrointestinales, renales, musculoesqueléticas, dermatológicas, neurológicas y psiquiátricas) que tienen en común la infiltración progresiva de los tejidos por células linfocitarias y plasmocitarias1,2, pero difieren en severidad, pronóstico, morbilidad y mortalidad. Asimismo, el SSp es llamado por algunos autores «la chaperona de la autoinmunidad» por su presencia concomitante con otras enfermedades autoinmunes —artritis reumatoide (AR), lupus eritematoso sistémico (LES), esclerosis sistémica— dentro del fenómeno de la poliautoinmunidad3,4.
Respecto a su epidemiología, en Latinoamérica y Colombia se estima que la prevalencia ronda entre el 0,08% y el 0,28% de la población mayor de 18 años5,6. Es una enfermedad de mayor incidencia en población femenina, con una edad de presentación más temprana en comparación con la descrita en cohortes europeas y una mayor incidencia de manifestaciones extraglandulares, dentro de las cuales las más frecuentes son la acidosis tubular renal, el compromiso articular y en el sistema nervioso central (SNC)7.
Con relación a las manifestaciones en el SNC, el problema surge de la falta de consensos o definiciones, a diferencia de lo que existe en el LES. Esto, sumado al amplio espectro de posibles manifestaciones neuropsiquiátricas, dificulta el diagnóstico y el apropiado tratamiento8. Por ello, el objetivo de esta revisión es hacer un recuento desde la visión de las diferentes especialidades involucradas —neurología, psiquiatría y reumatología— con el fin concientizar en torno a la búsqueda activa de dichas manifestaciones, la importancia de su abordaje multidisciplinario y el tratamiento orientado a ellas.
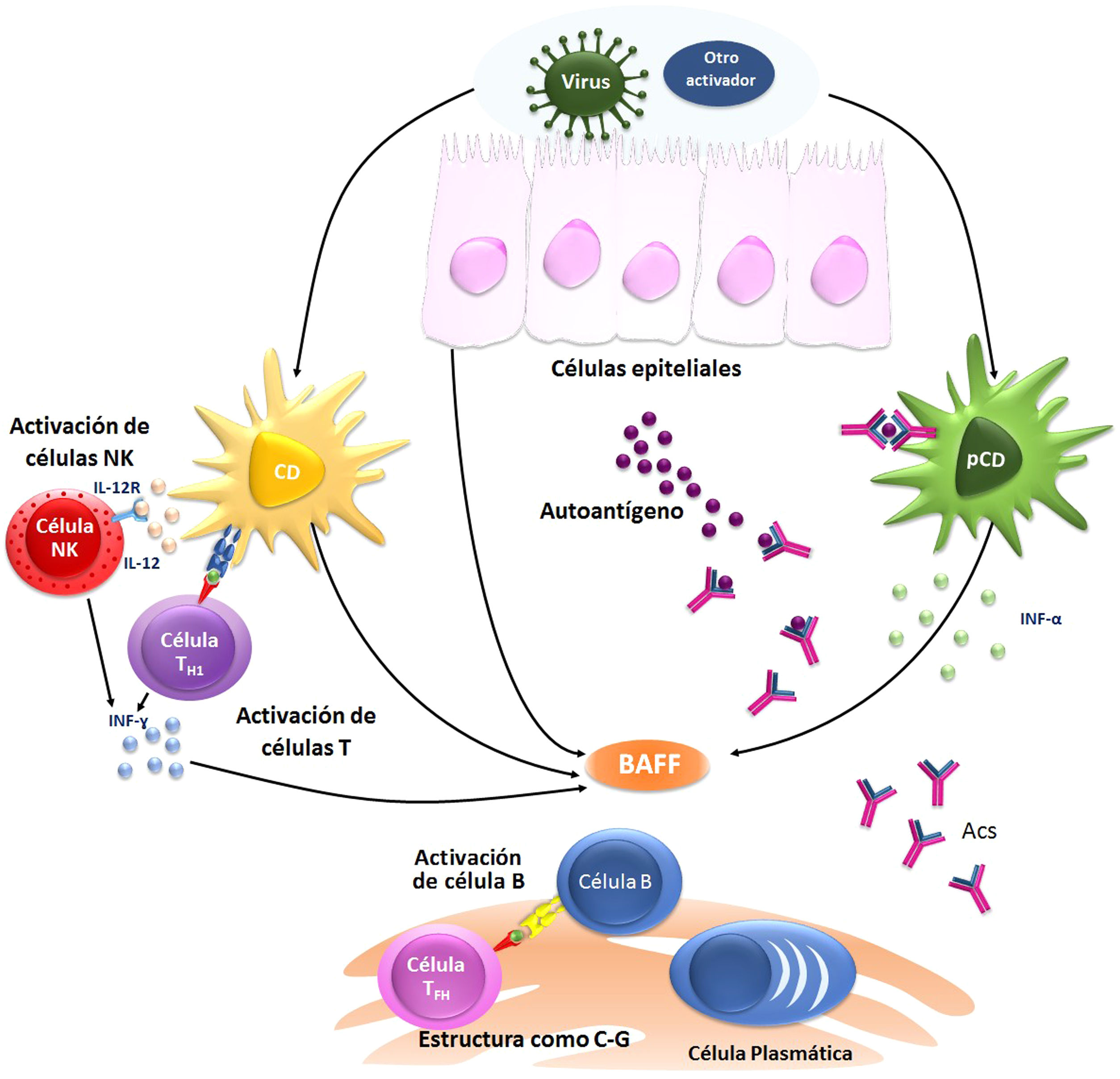
FisiopatologíaEn el SSp se conoce parte de los mecanismos inmunológicos que se ven alterados y que llevan al compromiso de las glándulas exocrinas, donde la activación de inmunidad innata potencia la respuesta adaptativa (fig. 1). La activación de la respuesta Th1 por medio de linfocitos T CD4+ y células Th17 promueve la formación de acúmulos linfocitarios que originan nuevos centros germinales de células autorreactivas, lo cual es potenciado por la producción del factor activador de células B (BAFF), la activación de las células plasmáticas y la producción de autoanticuerpos que perpetúan la inflamación y el daño tisular. Sin embargo, el origen y los mecanismos por los que se desencadena la inflamación local y sistémica que conllevan las manifestaciones glandulares y extraglandulares no está del todo esclarecido9,10.

En fecha reciente se han planteado algunas hipótesis a partir de los hallazgos histológicos y de las descripciones de autopsias de algunos pacientes con compromiso neuropsiquiátrico por SSp9,10. Una primera hipótesis plantea que hay infiltración de los tejidos del SNC por células mononucleares, y esto se fundamenta en los hallazgos en líquido cefalorraquídeo de pacientes con afectación neurológica que evidencian pleocitosis de predominio mononuclear con niveles elevados de inmunoglobulina G, lo que soporta la migración linfocitaria y de plasmocitos autorreactivos al SNC11. Además, en autopsias de pacientes con neuropatía sensitiva se ha documentado infiltración de linfocitos CD8+ y disminución en el número de neuronas en la raíz del ganglio dorsal12.
La segunda hipótesis plantea un daño endotelial por complejos inmunes y activación del complemento, en el cual existiría una relación con la presencia de autoanticuerpos de tipo anti-Ro y manifestaciones en otras localizaciones como vasculitis necrotizante en la piel, miositis y neuropatías periféricas13. La tercera hipótesis sugiere un componente vasculopático que se sustenta en hallazgos histológicos que documentan fenómenos isquémicos y hemorrágicos relacionados con compromiso del pequeño vaso por infiltración mononuclear (angeítis)14.
Estas tres hipótesis podrían no ser excluyentes, sino complementarias, lo cual explicaría la gran variedad de manifestaciones y también los diferentes abordajes terapéuticos. El reto surge de que la presencia de factores predisponentes que no son excluyentes entre sí en un individuo con una susceptibilidad genética determinada y, finalmente, una pérdida de la tolerancia inmunológica llevarían a la producción de autoanticuerpos antineuronales y al daño del sistema neuroinmunoendocrino, lo que causaría manifestaciones algunas de ellas potencialmente graves debido al SSp15–17.
Sistema nervioso centralDentro de las manifestaciones neurológicas del SSp solamente del 2 al 25% corresponde a compromiso del SNC18. De este grupo de manifestaciones, la seropositividad para los anticuerpos anti-SSA (Ro) se correlaciona de forma directa con el tipo de compromiso en SNC y es un predictor de severidad19. Este compromiso se puede dividir según su etiología, localización y manifestaciones clínicas asociadas (tabla 1), como se describe a continuación:
Manifestaciones neurológicas en el sistema nervioso central del síndrome de Sjögren
| + ACV - vasculitis |
| + Desórdenes del espectro de NMO |
| + Esclerosis múltiple-like |
| + Trastornos del movimiento |
| + Meningitis aséptica |
| + Paquimeningitis hipertrófica |
| + Manifestaciones cerebelosas no desmielinizantes |
ACV: ataque cerebrovascular; NMO: neuromielitis óptica.
El ataque cerebrovascular (ACV) debido a SSp es una manifestación infrecuente dentro de las manifestaciones focales neurológicas centrales de la enfermedad. Por ello, es necesario descartar otras etiologías o la poliautoinmunidad con LES o síndrome antifosfolípidos. En caso de confirmarse su relación con el SSp puede seguir un territorio vascular, cuyo origen probable se relaciona con el compromiso vasculopático secundario a la infiltración celular inflamatoria directa sobre el vaso sanguíneo, lo que se ha asociado con autoanticuerpos en SNC como anti-Ro20,21. Algunas de estas lesiones vasculares serán de pequeño vaso y de ellas la mayoría podría ser de curso asintomático o con lenta progresión de los síntomas, principalmente de tipo subcortical, y se presentarán como lesiones hiperintensas periventriculares en la resonancia magnética (RM) cerebral que simulan la leucoencefalopatía microangiopática vista en pacientes hipertensos, con factores de riesgo vascular o migraña22,23.
De acuerdo con su localización, estas lesiones pueden ocasionar diferentes síntomas: en el caso de presentarse en arterias principales cerebrales y su territorio motor o sensitivo primario, simularán un ACV de gran vaso de etiología tromboembólica; si se presentan en áreas elocuentes del lenguaje pueden desarrollarse parafasias o incluso afasia; si comprometen el cerebelo se desarrollará vértigo e inestabilidad; si se compromete el tallo cerebral habrá alteración de los movimientos oculares conjugados; si hay compromiso del territorio gangliobasal se presentarán movimientos anormales o parkinsonismo. Aunque menos frecuente, también puede haber compromiso de la médula espinal con lesiones de tipo vasculítico y desarrollo de síndromes medulares según la localización. Asimismo, pueden presentarse crisis convulsivas si cualquiera de estas lesiones tiene disposición cortical24–26.
Para el diagnóstico se realiza una RM cerebral que mostrará restricción de la señal en las secuencias de difusión que corresponden a las áreas de isquemia, las cuales serán hiperintensas en las secuencias de recuperación de la inversión atenuada de fluido (FLAIR, fluid attenuated inversion recovery) y T2. También, en estudios angiográficos se podrán apreciar áreas de estenosis arteriales segmentarias que corresponderán a vasculitis. Dichos estudios pueden ser normales, de acuerdo con el grado de actividad y severidad de la enfermedad. También es de utilidad la realización de punción lumbar, la cual mostrará pleocitosis leve con elevación de proteínas como manifestación de la inflamación27.
Siempre que se encuentren lesiones isquémicas cerebrales, el primer paso será llevar a cabo un estudio de factores de riesgo cardiovascular completo que descarte causas tratables e intervenibles, a pesar de que el paciente ya tenga diagnóstico de alguna enfermedad inmunomediada. Sin embargo, este estudio debe ser rápido, ya que, en caso de descartarse las etiologías aterotrombótica y cardioembólica, además de confirmarse la presencia de vasculitis de SNC, hay reportes de inicio temprano de esteroide a altas dosis con disminución de la recurrencia de los eventos. No obstante, las secuelas neurológicas y funcionales dependerán de la localización y la severidad de la lesión dentro del SNC19,24,25.
Desórdenes del espectro de la neuromielitis óptica (NMO)La neuromielitis óptica (NMO) es una entidad autoinmune del SNC caracterizada clásicamente por la presencia de anticuerpos de tipo inmunoglobulina G en suero contra la acuaporina 4 (AQP4). La AQP4 está presente en el SNC, en los podocitos de los astrocitos a nivel subependimario, dando un compromiso característico de localización axial, periependimario y centromedular28.
A lo largo de la historia ha existido controversia en torno a la NMO como manifestación del SSp en el SNC, dado que hay expresión de otras AQP en glándulas salivales y otros órganos comprometidos en el SSp; sin embargo, diversos estudios realizados hasta la fecha han encontrado que los AQP4 se encuentran solamente en pacientes con SSp que tienen algún desorden del espectro de la NMO, lo que sugiere que esta no es una manifestación específica del SSp en el SNC, sino que se comporta como una enfermedad autoinmune independiente. Por tal razón, trabajos como los de Birnbaum et al y Wingerchuk et al. han contribuido a sustentar que la presencia de AQP4 apoya la coexistencia de dos entidades autoinmunes bajo la teoría de la poliautoinmunidad28-30.
Dentro de las manifestaciones clínicas de la NMO se encuentran:
- •
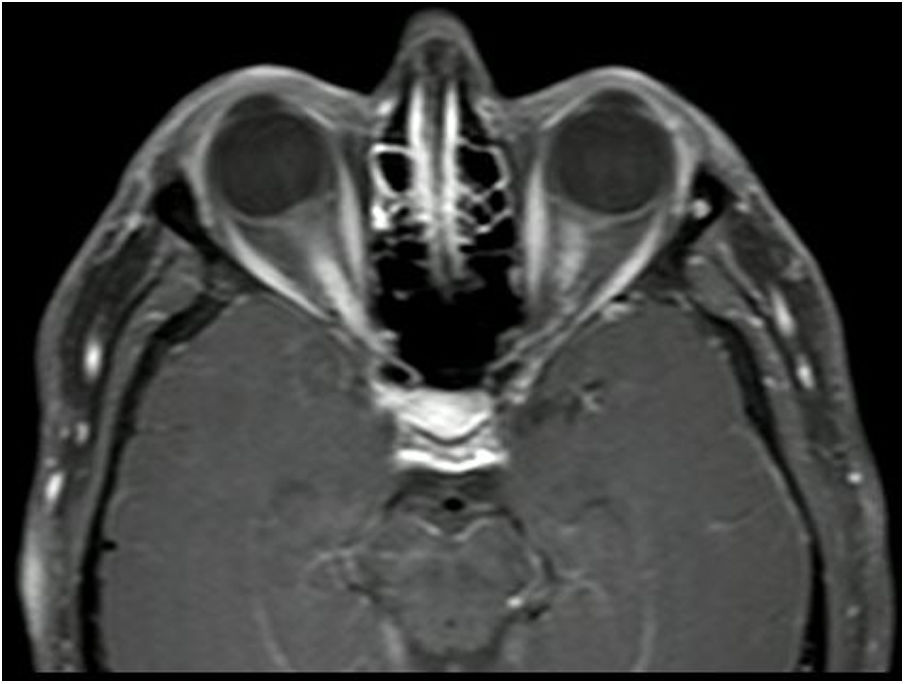
Neuritis óptica (figs. 2 y 3), que suele ser extensa, con compromiso del quiasma óptico o más del 50% de la longitud del nervio óptico y con un compromiso de moderado a severo de la agudeza visual.
- •
Las lesiones medulares por NMO son longitudinalmente extensas, es decir, comprometen tres o más segmentos medulares contiguos, y transversas o centromedulares, por lo que generan paraparesia o cuadriparesia asociada con síndromes sensitivos medulares, según el nivel de la lesión28.
- •
Fosa posterior. Puede presentarse como un síndrome de área postrema (hipo o vómito), síndrome de tallo cerebral (alteraciones motoras o sensitivas apendiculares o de movimientos oculares) y lesiones diencefálicas (narcolepsia). Pueden existir también lesiones cerebelosas que se presentan con ataxia, disartria o incoordinación apendicular31,32.
- •
En el 2017 se describió el caso de una paciente con una extensa lesión pontina y occipital que generó cuadro de ceguera cortical y encefalopatía. Esta paciente había presentado tres episodios de mielitis aguda en años previos. Se hace diagnóstico de NMO por seropositividad de anticuerpos anti-AQP4 en suero, así como de SSp por cumplir criterios ACR/EULAR33.
- •
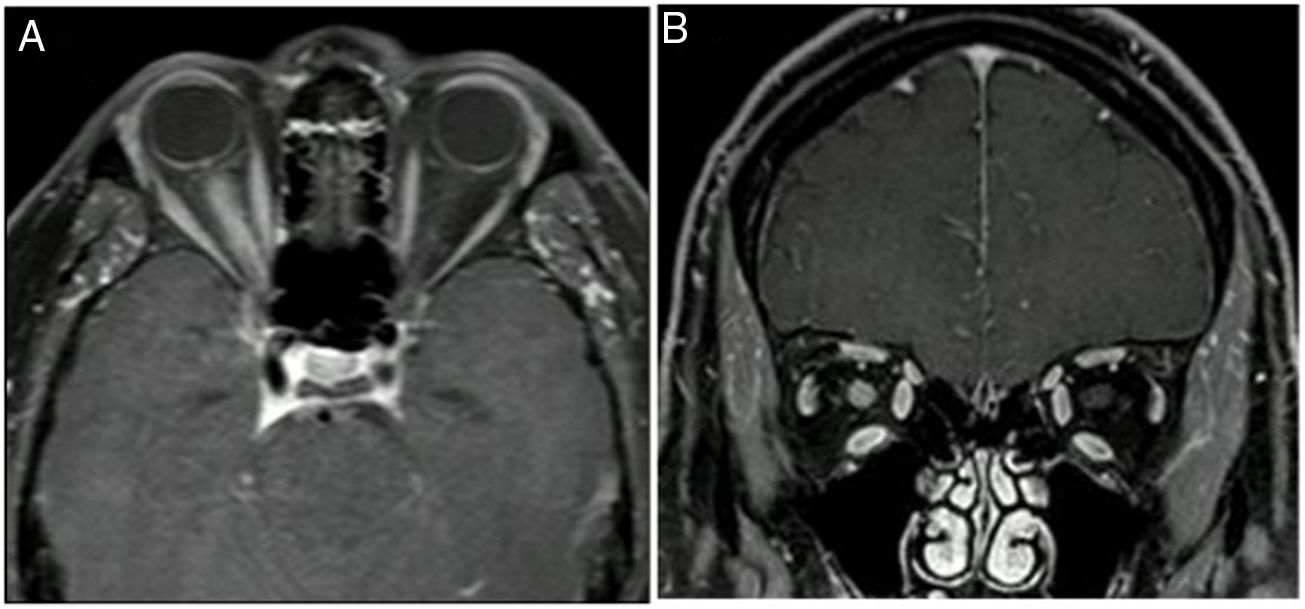
Las lesiones cerebrales tienden a ser confluentes y mal delimitadas, a nivel subcortical, periependimarias o de la sustancia blanca profunda, siguiendo el trayecto del tracto corticoespinal y comprometiendo la cápsula interna y los pedúnculos cerebrales. También se generan lesiones extensas, difusas y edematosas del cuerpo calloso. Las manifestaciones clínicas de las lesiones cerebrales se presentarán de acuerdo con su localización. Incluso puede haber lesiones asintomáticas. Hay un caso reportado en la literatura sobre una paciente que presentaba una lesión con efecto de masa en SNC, con gran edema como componente principal, que posteriormente fue diagnosticada de SSp, manejada con ciclofosfamida, y que luego desarrolló una neuritis óptica con anticuerpos anti-AQP4 en suero positivos, por lo que se le diagnosticó NMO34.

 y corte coronal (B).")
El diagnóstico de NMO requiere una evaluación metódica que considere diagnósticos diferenciales. Dicha evaluación debe tener en cuenta los síntomas clínicos descritos previamente, junto con neuritis y mielitis, con la presencia de anticuerpos IgG anti-AQP435.
El pronóstico no es bueno. Debido a las lesiones que se presentan puede haber compromiso severo de la visión y limitación funcional severa de la marcha por los síndromes medulares, lo cual puede mejorar desde que haya un adecuado tratamiento inmunomodulador. De la misma manera, el compromiso funcional dependerá de la localización de las lesiones, su severidad y el tratamiento oportuno de estas.
Esclerosis múltiple-LikeLa esclerosis múltiple (EM) es la enfermedad desmielinizante inflamatoria crónica más frecuente del SNC y la que más genera discapacidad en adultos jóvenes36. Se han descrito múltiples casos de lesiones similares a la EM en pacientes con SSp que inicialmente se consideraban una manifestación directa de la entidad en el SNC. Sin embargo, y de forma similar que en la NMO, se sabe que estos pacientes cumplen criterios clínicos y radiológicos para el diagnóstico de EM como entidad independiente del SSp. Además, es importante resaltar que muchos de los pacientes con lesiones desmielinizantes del SNC y SSp no cumplen criterios de McDonald 201737 para diagnóstico de EM, en cuyo caso siempre se deben buscar diagnósticos diferenciales como los desórdenes del espectro de NMO. La prevalencia de la EM en el SSp es baja y se compara con la de la población general, a diferencia de la NMO, cuya prevalencia es mayor en los pacientes con SSp33,38.
La EM se caracteriza clínicamente por lesiones medulares o cerebrales que pueden ser periventriculares, yuxtacorticales o corticales, e infratentoriales, así como también de nervio óptico. Los síntomas, dependiendo de su localización, pueden ser alteraciones sensitivas, hemiparesia o monoparesia, neuritis óptica o paraparesia con nivel sensitivo medular.
Para el diagnóstico de EM se debe demostrar diseminación en espacio y en tiempo en RM cerebral, con criterios de imagen ya definidos, o la presencia de bandas oligoclonales específicas en LCR. Una lesión desmielinizante única o atípica, con bandas oligoclonales positivas, sin cumplir criterios McDonald 2017, no hace diagnóstico de EM37,39.
Trastornos del movimientoSe definen como un exceso o enlentecimiento de los movimientos voluntarios o automáticos. A pesar de que se conoce que hay una interrelación de los circuitos cerebrales con diferentes zonas del cerebro, los ganglios basales son la estructura principal asociada al control del movimiento. En el SSp estos desórdenes del movimiento pueden ser explicados por lesiones vasculíticas gangliobasales, por una lesión desmielinizante asociada a la NMO, o bien por un daño directo mediado por anticuerpos antineuronales sin lesión estructural visible en RM cerebral 40,41.
Clínicamente, estos trastornos se pueden presentar como corea, la cual resulta ser una presentación infrecuente. Los reportes de caso asociados a SSp han sido de corea generalizada, ya sea con RM cerebral normal, asociando la etiología a daño directo por anticuerpos antineuronales, o con lesiones gangliobasales bilaterales y ligero realce con gadolinio en RM cerebral, caso en el cual la etiología se asocia a vasculitis. En los dos casos el manejo fue con esteroides, en uno de ellos con azatioprina asociada, con lo cual hubo mejoría de los síntomas40,41.
Existen reportes de caso de distonía asociada a SSp como manifestación en SNC, una manifestación igualmente infrecuente. En uno de los casos la presentación de la distonía fue focal en la mano izquierda, y ninguno de los casos tenía anormalidades en la RM cerebral. Sin embargo, el caso de la distonía focal presentaba disminución en la captación del estriado derecho en el SPECT. El manejo inicial en los dos casos fue con esteroide a altas dosis, en uno con refractariedad a la azatioprina, por lo cual se cambió el manejo a ciclofosfamida y los síntomas mejoraron42,43.
Se han encontrado varios reportes de caso de parkinsonismo atípico asociado a SSp que se presenta con bradicinesia, inestabilidad postural, cambios de la marcha, rigidez y temblor. Dos de estos casos tuvieron RM cerebral normal, uno presentaba hipometabolismo estriatal y cortical por tomografía por emisión de positrones (PET) y otros presentaron lesiones difusas, hiperintensas en T2, ganglios basales y de la sustancia blanca periventricular. Recibieron manejo con levodopa y esteroides, con parcial respuesta clínica, algunos de ellos sin ninguna mejoría44.
Meningitis asépticaLa meningitis aséptica es frecuente en los pacientes con SSp, los cuales clínicamente se presentan con cefalea, pueden tener signos meníngeos y síntomas prodrómicos virales. Además, pueden o no presentarse con fiebre baja. En el LCR se encuentra pleocitosis linfocitaria leve a moderada con proteínas elevadas, sin aislamiento infeccioso en el LCR, y se cree que este hallazgo es secundario a la inflamación de los vasos meníngeos. La RM cerebral con gadolinio suele ser normal, o puede tener pequeñas áreas de hiperintensidad cortical o áreas de vasculitis. El tratamiento es de soporte, analgesia y una adecuada modulación de la actividad de la enfermedad18,26,45.
Paquimeningitis hipertróficaLa paquimeningitis hipertrófica es una rara entidad inflamatoria caracterizada por engrosamiento focal o difuso de la duramadre. Se reconocen dos formas: la primaria o idiopática, cuando no hay una causa identificable, y la secundaria, cuando se logra identificar la etiología. Dentro de las causas más frecuentes se encuentran la malignidad, las infecciones crónicas y la infiltración por enfermedades inmunomediadas. En un estudio japonés en el que se evaluaron las causas de paquimeningitis hipertrófica en una cohorte de 149 pacientes, se encontró que la causa más común es la idiopática (44%), seguida de la vasculitis asociada a ANCA (34%) y la enfermedad por IgG4 (8,8%). Solamente el 1,3% de los casos correspondió al SSp46.
Se cree que la causa directa es la infiltración inflamatoria de linfocitos y células plasmáticas, lo que genera engrosamiento de la duramadre y ha sido puesto en evidencia en muestras histológicas de biopsias meníngeas de pacientes aún sin diagnóstico establecido. La clínica de la paquimeningitis hipertrófica comprende un amplio espectro de signos y síntomas que pueden ir desde cefalea, mareo, vértigo o hipertensión endocraneana, hasta lo relacionado con el engrosamiento o la infiltración de estructuras adyacentes como el II, V y VIII nervios craneales, entre otros. Para el diagnóstico se requiere engrosamiento y realce de la duramadre en la RM cerebral, ya sea focal en base de cráneo, o de forma difusa. En algunos pacientes se encuentra primero el hallazgo radiológico, y en los estudios complementarios hay evidencia serológica de SSp47,48.
El pronóstico en general es bueno, las secuelas dependerán del grado de compromiso compresivo sobre estructuras sensibles como los nervios craneales. La cefalea, el síntoma más común, mejora satisfactoriamente en la mayoría de los casos desde el inicio del tratamiento47,48.
CerebeloLas manifestaciones cerebelosas de tipo no desmielinizante en el SSp son poco frecuentes, pero altamente incapacitantes. Se presentan clínicamente de forma insidiosa y con deterioro progresivo, generan ataxia de la marcha, disartria, incoordinación apendicular, nistagmus o vértigo. Algunos de estos pacientes debutan con la clínica cerebelosa y posteriormente se hace el diagnóstico de SSp31,32.
Puesto que la etiología no es desmielinizante, la RM cerebral puede ser normal o mostrar atrofia cerebelosa en diferentes grados, lesiones inespecíficas de sustancia blanca o áreas de necrosis. Sin embargo, en caso de ser normal y persistir con alta sospecha clínica, se recomienda la realización de PET, en el cual se podrá observar disminución de la captación en los hemisferios cerebelosos31,32.
No hay un consenso sobre el tratamiento de estas manifestaciones, aunque se recomienda el manejo con esteroides a altas dosis para la fase aguda, así como una adecuada inmunomodulación de la enfermedad de base con las diferentes opciones terapéuticas disponibles31,32.
Sistema nervioso periféricoEl compromiso del sistema nervioso periférico es una manifestación extraglandular común del SSp primario49. De estas manifestaciones la más prevalente es la neuropatía sensitiva50. Por lo tanto, dentro de las formas de compromiso se encuentran las neuropatías periféricas en sus diferentes presentaciones y las miopatías inflamatorias (tabla 2).
Manifestaciones neurológicas en el sistema nervioso periférico del síndrome de Sjögren
| + Neuropatía sensitivomotora |
| + Neuropatía sensitiva |
| + Neuropatía de fibra pequeña |
| + Mononeuritis múltiple |
| + Ganglionopatía |
| + Polirradiculoneuropatía desmielinizante inflamatoria crónica |
| + Neuropatía autonómica |
| + Neuropatía craneal |
| + Enfermedad de neurona motora |
| + Miopatía |
La frecuencia de aparición de neuropatía periférica en pacientes con SSp es variable. Según distintos autores, oscila entre 2 y 60%51–53. La neuropatía periférica puede ser incluso la presentación inicial de la enfermedad, llegando a ser el primer síntoma hasta en el 25% de los pacientes con SSp54, y precediendo al síndrome seco por un intervalo de hasta de 24 meses en promedio55. De estas manifestaciones, la más frecuente es la neuropatía sensitivomotora56.
El principal mecanismo fisiopatológico implicado en la mayoría de las neuropatías asociadas a SSp es la vasculitis, excepto por las ganglionopatías, las cuales involucran una infiltración linfocítica del ganglio de la raíz dorsal y se sugiere un posible rol de anticuerpos antineuronales57. En cuanto a las vasculitis, es común encontrar cambios como infiltración inflamatoria de linfocitos T CD8+ y necrosis de la vasa nervorum. En el nervio periférico predomina la degeneración axonal sobre la desmielinización, la cual puede ser focal o multifocal. Es común encontrar pérdida de fibras mielinizadas y de fibra pequeña56.
En general, la aparición de otras manifestaciones secundarias a SSp parece ser mayor en presencia de anticuerpos anti Ro/SSA o anti La/SSB, puesto que son indicativos de un perfil inmunológico activo58. En la neuropatía periférica asociada a SSp estos anticuerpos suelen ser negativos59, pero cuando se observa positividad, esta es más frecuente en pacientes con compromiso del sistema nervioso periférico que en aquellos con compromiso del sistema nervioso central60.
La presencia de la neuropatía periférica se considera un factor de mal pronóstico en el SSp, en especial en los casos de mononeuritis múltiple y polineuropatía sensitivomotora, debido a que se correlaciona con peor calidad de vida, peor pronóstico y otras manifestaciones extraglandulares como glomerulonefritis o asociación con neoplasias hematolinfoides18,61,62.
A continuación se discuten los aspectos relevantes de cada tipo de neuropatía asociada al SSp.
Neuropatía sensitivomotoraSu frecuencia en pacientes con SSp oscila entre un 20 y un 30%63, y suele tener un curso lento e insidioso. La mayoría inicia con afectación sensitiva, usualmente parestesias, que predominan en los miembros inferiores y con frecuencia son distales y simétricas. Los síntomas motores muchas veces son discretos, en forma de debilidad leve, y aparecen de manera gradual con la progresión de la enfermedad. El compromiso motor es simétrico, involucra músculos distales como los extensores del pie y es común encontrar hipo o arreflexia. La asociación con otras formas de compromiso extraglandular es más frecuente y marcada en comparación con la polineuropatía sensitiva, y por lo general se acompaña de púrpura, vasculitis, hipocomplementemia (C4) y crioglobulinemia64. En las neuroconducciones se observa polineuropatía con degeneración axonal como mecanismo primario, y se afectan sobre todo las fibras sensitivas de los miembros inferiores. En la biopsia de nervio periférico se evidencia un adelgazamiento de las fibras mielinizadas y degeneración axonal63.
Neuropatía sensitivaSu curso es insidioso y de instauración crónica. Se caracteriza por parestesias simétricas distales que pueden o no acompañarse de dolor urente. Los síntomas son más comunes, de mayor intensidad en miembros inferiores, y solamente el 20% tiene compromiso de miembros superiores18. Suele haber hipo o arreflexia. Las neuroconducciones evidencian un patrón axonal simétrico que compromete fibras sensitivas, y entre los hallazgos anatomopatológicos puede evidenciarse degeneración axonal18.
Neuropatía de fibra pequeñaOcurre por el daño de las fibras finas A-delta mielinizadas, o de las fibras C no mielinizadas, las cuales conducen la información termoalgésica. Se considera de alta prevalencia en cohortes de pacientes con SSp, se estima que su frecuencia es del 5-10%. Sin embargo, no es fácil de establecer debido a que no se dispone de las herramientas para diagnosticarla ya que las neuroconducciones suelen ser normales65. La clínica es de instauración gradual y de curso subagudo a crónico. Se caracteriza por ser una neuropatía muy dolorosa, urente, que puntúa al menos 5 en la escala análoga del dolor18, frecuentemente distal y simétrica, y se asocia con hipoestesia nociceptiva y térmica en forma de zonas parcheadas.
Por lo general, este tipo de neuropatía no compromete la vibración ni la propiocepción, pero al progresar puede generar alteración de la sensibilidad profunda por compromiso de fibras más gruesas63. Los reflejos musculotendinosos y la vibración se conservan, los estudios de neuroconducción suelen ser normales, y en ocasiones se pueden encontrar alterados los potenciales evocados somatosensoriales. Es la neuropatía que con mayor frecuencia se asocia con anticuerpos antinucleares (ANAS) positivos66. El diagnóstico se confirma con biopsia cutánea, la cual muestra una disminución de la densidad de fibras nerviosas intraepidérmicas e infiltración linfocítica de predominio CD8+ perineuronal65.
Otras alternativas de estudios diagnósticos exploradas recientemente son la determinación de umbrales cálidos y fríos (WDT, CDT por sus siglas en inglés), el registro de potenciales evocados por láser, el registro de respuestas simpáticas en piel y la conductancia electroquímica en piel. Sin embargo, aún no se ha determinado qué tan precisas son estas pruebas en comparación con el patrón de oro67. El tratamiento es principalmente sintomático; entre los analgésicos neuromoduladores que se utilizan están los tricíclicos, la duloxetina, la gabapentina y la pregabalina. No obstante, algunos autores contraindican el uso de amitriptilina por sus efectos anticolinérgicos, pues estos pueden empeorar el síndrome seco. Por este motivo, se recomienda preferiblemente el uso de tricíclicos de aminas secundarias como la nortriptilina y la desipramina (tienen menor efecto anticolinérgico), así como el uso de gabapentina y pregabalina, como agentes de primera línea, y como segunda línea los opioides56,68,69.
Mononeuritis múltipleConsiste en un daño asimétrico, simultáneo o consecutivo de al menos dos nervios de raíces no contiguas. Su prevalencia en el SSp varía según los distintos autores, oscila entre un 12 y un 50%70–72 y se considera que con un diagnóstico oportuno y un tratamiento temprano es la neuropatía que presenta mejor respuesta a la terapia73. El cuadro clínico tiene una instauración aguda a subaguda, longitud-dependiente, iniciando con parestesias y disestesias distales en miembros inferiores. Una de las manifestaciones iniciales es el pie caído56, y por debilidad de los músculos dorsiflexores, algunos pacientes pueden presentar marcha en estepaje. De manera ocasional se ven afectados el nervio trigémino y los nervios intercostales. Puede haber dolor profundo en la parte proximal de la extremidad afectada o parestesias dolorosas en el territorio del nervio sensitivo afectado, o, por el contrario, puede ser indoloro y predominar la debilidad de la extremidad.
Por lo general, se asocia con vasculitis cutánea y crioglobulinemia, motivo por el cual algunos autores consideran que los pacientes con mononeuritis múltiple en el contexto del SSp deben ser evaluados para crioglobulinemia56, y debido a que involucra un proceso vasculítico, puede acompañarse de síntomas constitucionales. También pueden verse elevadas la velocidad de sedimentación globular (VSG) y la proteína C reactiva.
En los estudios neurofisiológicos se encuentra una reducción del potencial de acción sensitivo y compuesto, daño axonal y pseudobloqueos, estos últimos correspondientes a áreas de isquemia del nervio. Suele ser necesaria la realización de una biopsia para confirmar los hallazgos de vasculitis. Dentro de los hallazgos anatomopatológicos se puede encontrar una vasculitis asociada con necrosis fibrinoide. Asimismo, puede verse una disminución de la fibra pequeña y de la fibra gruesa, además de degeneración axonal activa, y en áreas de isquemia del nervio se puede observar necrosis de la vasa nervorum con infiltración de linfocitos T y macrófagos. El pronóstico depende del diagnóstico y del tratamiento temprano con inmunosupresores y glucocorticoides en la fase aguda66,74,75.
Ganglionopatía/neuronopatía atáxica sensitivaInvolucra un daño selectivo del ganglio de la raíz dorsal. Los síntomas generalmente preceden el diagnóstico de SSp, y es la neuropatía que menos se asocia con otras manifestaciones extraglandulares76. Puede haber compromiso de todas las modalidades sensitivas, pero usualmente las fibras que más se afectan son las gruesas de tipo 1a, que conducen señales de los husos musculares. Debido a esto, el signo encontrado con mayor frecuencia es la ataxia sensitiva, debido a una pérdida de la propiocepción y de la vibración, inestabilidad para la marcha secundaria y aumento del polígono de sustentación con signo de Romberg positivo. La inestabilidad secundaria para la marcha varía en severidad, al extremo de ocasionar dependencia completa para el desplazamiento.
Los pacientes cursan también con parestesias en las cuatro extremidades, pero de predominio en miembros superiores, inicialmente en dedos de manos y pies. Con el tiempo se torna simétrica y compromete toda la extremidad y el tronco. A diferencia de la mononeuritis múltiple y de la neuropatía sensitiva, se encuentra hipo o arreflexia, con ataxia sensitiva, pero sin debilidad muscular77,78. De forma anecdótica, se ha descrito el desarrollo de la artropatía de Charcot secundaria a neuronopatía sensitiva derivada de SSp79.
Puede observarse disminución o ausencia de los potenciales de acción de los nervios sensitivos, un patrón axonal asimétrico en las neuroconducciones63, potenciales somatosensoriales anormales y electromiografía normal. Los hallazgos en la resonancia magnética pueden variar desde no encontrar alteraciones hasta documentarse hiperintensidad en T2 de los cordones posteriores63. La biopsia revela pérdida de cuerpos neuronales e infiltración de linfocitos T en el ganglio de la raíz dorsal, así como reducción de fibras gruesas56. La ganglionopatía es una complicación severa, usualmente refractaria a tratamiento80.
Polirradiculoneuropatía desmielinizante inflamatoria crónica (CIDP)La CIDP es un hallazgo poco común en los pacientes con SSp18,75,81. Se caracteriza por una alteración sensitivomotora crónica progresiva con parestesias en guante y bota, ataxia sensitiva y debilidad muscular. En algunas ocasiones se presentan síntomas autonómicos como diarrea, hipohidrosis o alteraciones urinarias, y en la valoración se presenta hipo o arreflexia. En líquido cefalorraquídeo se evidencia disociación albúmino-citológica en el 80 a 90% de los pacientes18. Los estudios electrodiagnósticos demuestran una etiología desmielinizante68, mientras que en la biopsia de nervio periférico hay una pérdida de fibras mielinizadas y cambios leves a moderados en las no mielinizadas63.
Neuropatía autonómicaSe ha descrito una prevalencia alrededor del 6,3%55. Sin embargo, en una cohorte reciente de 154 pacientes con SSp se observó disfunción autonómica hasta en un 35,7%, así como un mayor índice de fatiga en la escala EULAR SSp Patient Reported Index82. Las distintas manifestaciones clínicas reportadas involucran pupila de Adie, hipotensión ortostática, hipo o anhidrosis de tronco y extremidades, y dolor abdominal con diarrea o estreñimiento. Dentro de los paraclínicos se pueden encontrar alteraciones en las pruebas de reflejos cardiovasculares, retraso en el vaciamiento gástrico y disfunción vesical.
En la gammagrafía con I-metaiodobenzilguanidina (I-MIBG) puede observarse una disminución en la captación de contraste en los casos de ortostatismo grave. Se ha reportado un caso de neuropatía autonómica autoinmune asociada con síndrome seco, con elevación de anticuerpos antirreceptor de acetilcolina83. Dentro de los tratamientos descritos se han empleado la fludrocortisona o el midodrine56; no se ha demostrado respuesta al tratamiento con IGIV o prednisolona72.
Pares cranealesLa disfunción de par craneal con mayor frecuencia asociada con SSp es la del nervio trigémino (V). Algunos autores plantean que esta quizá se deba a una infiltración inflamatoria del ganglio de Gasser84. Usualmente el compromiso es unilateral, debido a parestesias en las ramas inferiores, en particular la rama maxilar. También se han descrito hipoestesias faciales y disestesias en lengua63.
Así mismo, se ha descrito compromiso del nervio facial (VII) por paresia facial, del vestibulococlear (VIII) debido a hipoacusia y síndrome vestibular, y de los oculomotores (III, IV, VI) por diplopía60. Mori et al. describieron neuropatías de múltiples nervios craneales, entre ellos III, V, VI, VII, IX y XII en diversas combinaciones. Los pacientes pueden estar asintomáticos y evidenciarse su disfunción como hallazgo incidental en estudios neurofisiológicos72,85,86.
Enfermedad de neurona motoraSu presentación en concomitancia con SSp es anecdótica87–89. Clínicamente se expresa con debilidad, atrofia de músculos esqueléticos y fasciculaciones generalizadas, incluida la lengua. Entre los casos descritos no se encontraron signos de lesión de motoneurona superior. En las neuroconducciones de nervios motores se evidencia reducción de la amplitud del potencial de acción muscular compuesto y latencia prolongada o ausencia de onda F. La electromiografía muestra cambios por denervación aguda y fasciculaciones. Se describió mejoría después del tratamiento con corticoesteroides e inmunosupresores90.
MiopatíaAnte un síndrome de debilidad muscular en SSp, debe considerarse la posibilidad de enfermedad muscular inflamatoria como poliautoinmunidad con dermato y polimiositis dentro de las manifestaciones extraglandulares del SSp56. Clínicamente, el paciente con miopatía presenta debilidad simétrica de predominio proximal, normorreflexia y elevación de la CPK. De las miopatías inflamatorias, la miositis por cuerpos de inclusión (MCI) es la más comúnmente asociada a SSp91, con pocos beneficios clínicos y en pronóstico a pesar del uso de la terapia inmunomoduladora con glucocorticoide, metotrexate y azatioprina. Como resultado, se encuentra disminución de niveles séricos de CPK en ausencia de respuesta clínica92.
Otra de las entidades documentadas en SSp es la polimiositis por patología mitocondrial, la cual se diferencia de la MCI por presentar debilidad muscular de patrón simétrico y proximal y por presencia de deleciones en el ADN mitocondrial por PCR. En cuanto al tratamiento en asociación con SSp, el uso de metilprednisolona, metotrexate y suplemento de calcio presenta desenlaces no claros en mejoría clínica, con resultados favorables en algunas series, pero no sostenidos a largo plazo93.
PsiquiátricoAlteraciones afectivas y ansiedadEl SSp, debido a sus características de ser de curso crónico y debilitante, puede afectar el estado mental de los pacientes y producir en algunas ocasiones síntomas de depresión y ansiedad como síndromes predominantes que alteran de manera significativa la calidad de vida94.
La depresión tiene reportes de prevalencia más alta en estos pacientes que en la población general, con un amplio rango que va del 8,3 al 75,5%94,95. La presencia de ojo seco, con independencia de la patología subyacente, se ha asociado con una prevalencia tres veces mayor de depresión y ansiedad. Asociado con esto también se ha documentado un mayor índice de fatiga y disminución de la funcionalidad en actividades de la vida diaria, pero al comparar pacientes con SSp y pacientes con síntomas secos sin SSp se encuentra que la proporción de pacientes con síntomas depresivos y ansiosos tiende a ser similar, sin que se reporten diferencias entre ambos grupos96–98.
Otro impacto de la asociación SSp con depresión y ansiedad es la presencia de trastornos del sueño, disminución en la adherencia terapéutica, mayor tasa de desempleo, inestabilidad laboral, disminución de la productividad, discapacidad física y mayores costos en salud al comparar con pacientes con SSp sin síntomas depresivos56,94.
Frente a la expresión de síntomas depresivos, sus mecanismos no han sido claramente identificados y se postula un componente multifactorial que incluye factores intrínsecos y factores extrínsecos. Por un lado, se asocia con la presencia de pensamientos rumiantes mal adaptativos que median la relación entre la enfermedad y las emociones negativas. Desde la psiconeuroimunología, la depresión se ha asociado con elevación de citoquinas proinflamatorias, en tanto que desde el enfoque de personalidad y afrontamiento, las personas con un alto neuroticismo y bajo autodireccionamiento son más propensas a presentar síntomas depresivos95.
Alteraciones cognitivasEn el SSp pueden ocurrir alteraciones cognitivas. En tal sentido, algunos han sugerido que la aparición de fallas de este tipo se relaciona con un deterioro cognitivo leve97, asociadas con un posible componente multifactorial (dolor, síntomas depresivos, alteraciones del sueño y uso de medicamentos), endotelitis inmunomediada99 o infiltración directa del tejido encefálico con cambios inflamatorios que llevan a una disfunción en las vías fronto-subcorticales. De tal manera, se encuentra una correlación positiva entre las anormalidades en sustancia blanca, la hipoperfusión en áreas frontal, temporal, parietal, cíngulo e hipocampo y el funcionamiento cognitivo, en especial la disfunción ejecutiva97.
La disfunción se ha identificado en 44 a 50% de los pacientes, expresada por medio fallas en la atención, en la velocidad de procesamiento de la información, en las funciones ejecutivas, en la memoria visual y verbal (corto y largo plazo) y en la percepción visoespacial, lo que se soporta por un rendimiento menor en las pruebas del reloj, COWAT, PASAT, STROOP, SDLT, AVLT, BJLOT, RCFT y BNT97.
Blanc et al. publicaron una cohorte de 25 pacientes con SSp, con diferentes alteraciones cognitivas, en la que un hallazgo llamativo era la presencia de lesiones en sustancia blanca periventriculares con una correlación positiva entre la severidad de las alteraciones cognitivas y los hallazgos imagenológicos reportados100.
Los casos de trastorno neurocognoscitivo mayor son pocos, en su mayoría con alteraciones cortico-subcorticales (alteraciones vasculares o sinucleinopatias), y algunos casos de enfermedad de Alzheimer que se pueden asociar como una comorbilidad y no como una manifestación del SSp. No obstante, el trastorno neurocognoscitivo inducido por actividad autoinmune es posible en el SSp y por definición es reversible, por lo que debe hacerse un diagnóstico preciso y descartar la patología degenerativa primaria no asociada a SSp99.
Trastornos del sueñoLos cambios del patrón de sueño deben entenderse a través de la mirada del envejecimiento, pues hay cambios normales (tiempo total de sueño, latencia, profundidad, despertares nocturnos, somnolencia diurna, siestas diurnas, factores hormonales y de regulación de temperatura, entre otros) que deben ser tenidos en cuenta para ajustar la evaluación de las alteraciones del sueño de un paciente101. Las variaciones que están por fuera de los cambios esperables terminan volviéndose eventos patológicos que deben ser intervenidos.
El 23,7% de los pacientes con síntomas secos debido a SSp presenta un menor tiempo total de sueño, mayor somnolencia diurna, dolor, nocturia, fatiga física y mental, insomnio, síntomas ansiosos, aumento de la latencia del sueño, despertares nocturnos, síndrome de piernas inquietas, apnea obstructiva de sueño, lo que hace necesaria una evaluación del sueño como parte de su estudio95,102–106.
Como medidas terapéuticas se recomienda la terapia cognitivo-conductual, la humidificación nocturna y los spray salivales artificiales. Ante las alteraciones del sueño y su clara asociación con síntomas mentales y empeoramiento de la calidad de vida, la consulta a psiquiatría se vuelve necesaria, lo que convierte al SSp y a los síntomas secos en patologías multidisciplinarias. Asimismo, es de gran importancia recordar que el tratamiento farmacológico también puede ser responsable de exacerbar los síntomas sicca104.
Alteraciones en la personalidad y otros trastornos mentalesEn el contexto del SSp se han reportado algunos casos esporádicos de manifestaciones psiquiátricas inusuales tales como episodios psicóticos107 o trastorno obsesivo compulsivo con síntomas depresivos asociados (episodio depresivo mayor con síntomas mixtos), con resolución de la sintomatología mental luego del tratamiento inmunosupresor108. Lo anterior sugiere que la evaluación reumatológica debe incluir un enfoque hacia los síntomas mentales para poder detectarlos y hacer intervenciones tempranas que busquen mejorar la calidad de vida del paciente95.
En otro trabajo Milic et al., luego de evaluar los rasgos de personalidad de 105 pacientes con SSp mediante el inventario de personalidad NEO, encontraron una alta puntuación y mayor riesgo de neuroticismo, así como baja puntuación con bajo riesgo para extraversión, comparado con el grupo control109. El neuroticismo es una tendencia a experimentar emociones negativas y se asocia con una predisposición a la vulnerabilidad y el estrés psicológico110, lo que explica su relación con la percepción del dolor y por ende de las alteraciones en la calidad de vida. Estos resultados sugieren que los mecanismos de afrontamiento del estrés pueden predisponer a desarrollar síntomas afectivos en los pacientes que los presentan109, lo que resalta el carácter multifactorial de las manifestaciones mentales.
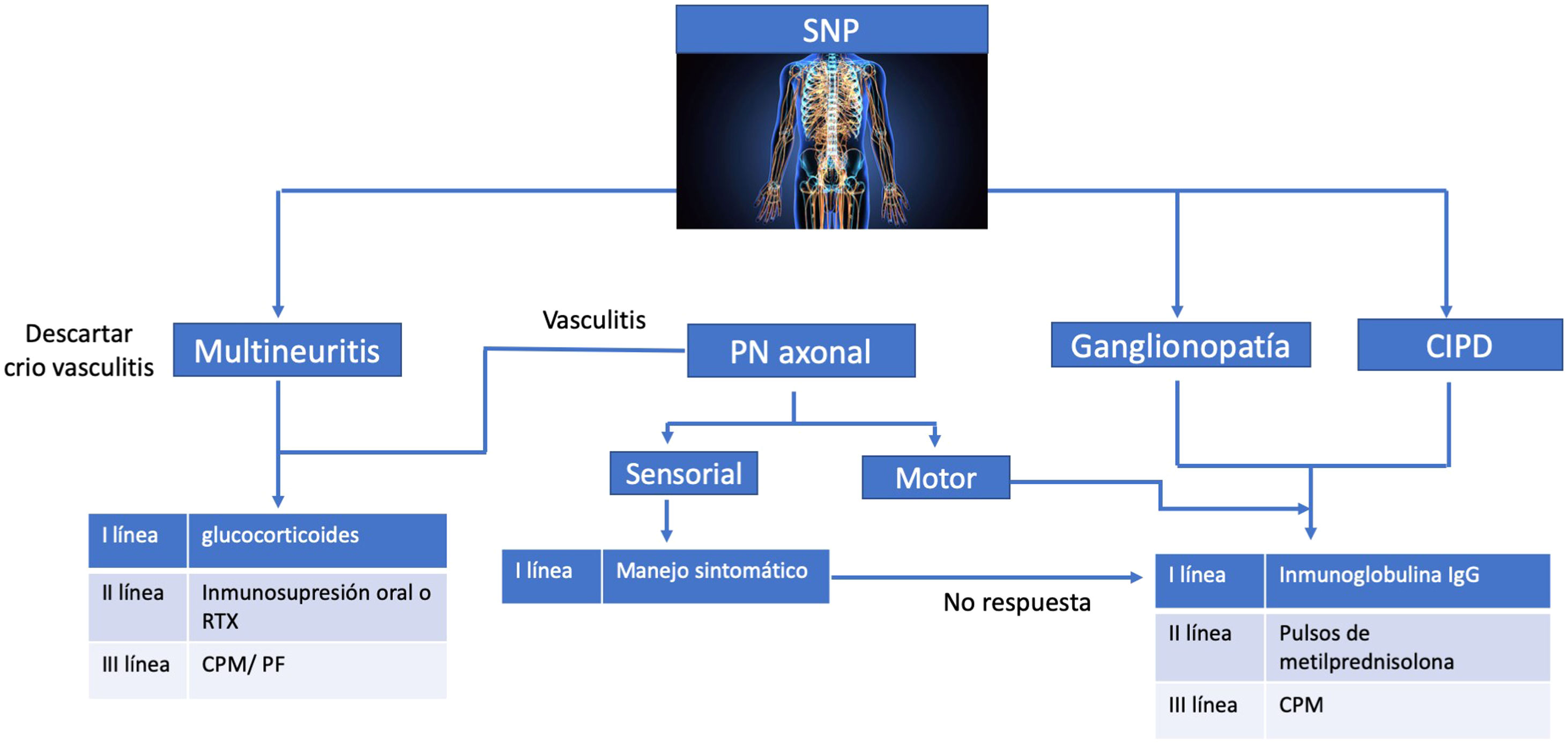
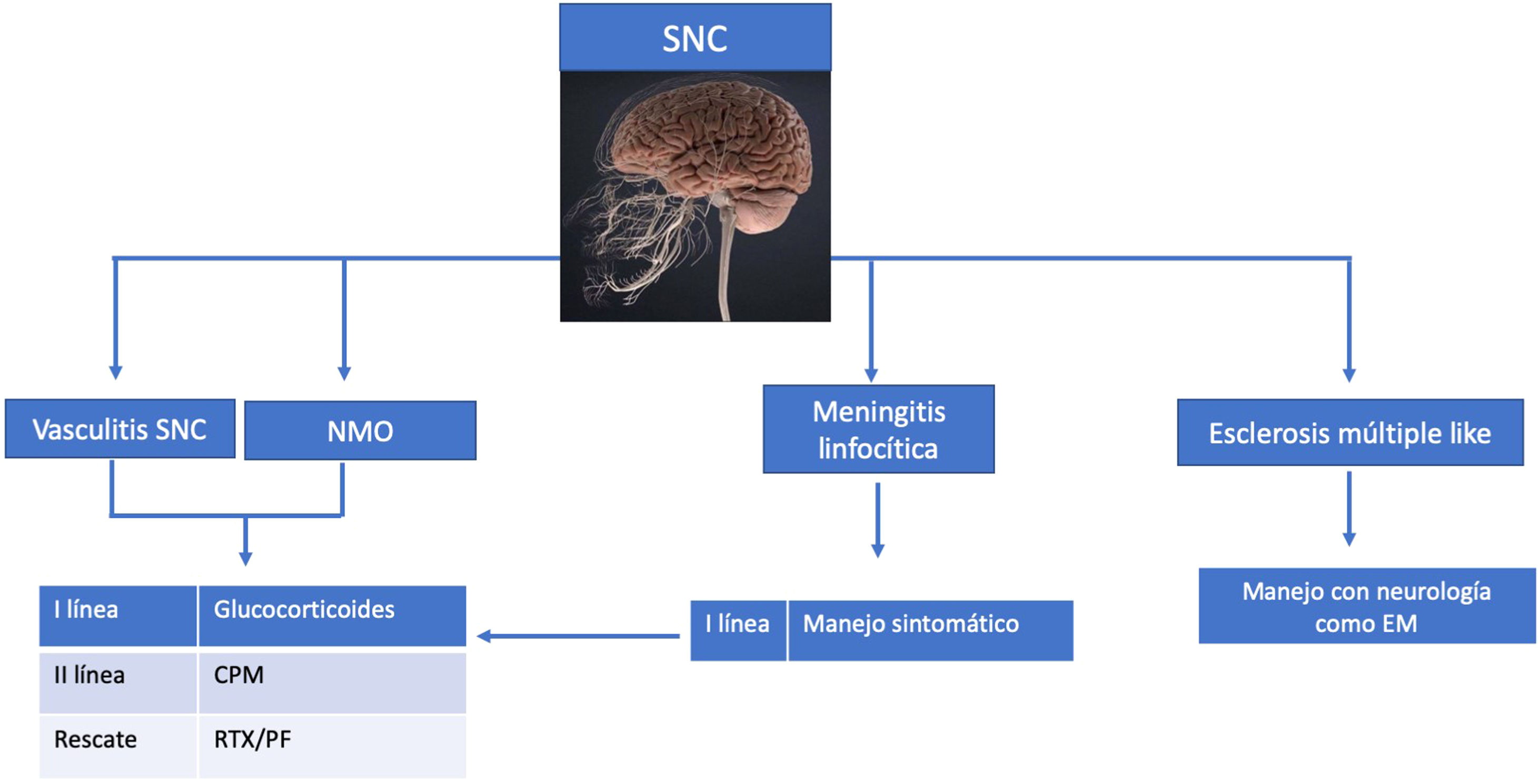
TratamientoEl SSp no ha sido considerado una entidad que suela poner en riesgo la vida, o que tenga gran morbilidad o mortalidad, pero existen condiciones apremiantes que obligan a un manejo inmunosupresor. Por tal motivo, en el año 2019 EULAR realizó un consenso de expertos acerca de cuál era la mejor evidencia disponible para el manejo de las manifestaciones extraglandulares, dentro de las cuales están incluidas las neurológicas, algunas de las cuales se han mencionado en estas líneas (figs. 4 y 5)111.

Manejo para compromiso sistema nervioso periférico, adaptado de las guías EULAR 2019120.

Manejo para compromiso sistema nervioso periférico, adaptado de las guías EULAR 2019120.
La utilización de la clinimetría, tan ampliamente difundida en la reumatología para entidades como la AR o el LES, en el SSp cada vez tiene mayor cabida con la utilización del EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI), que hace referencia a la alta actividad de la enfermedad, o del consenso de 2019 que resalta las neuropatías (ganglionopatía y poliradiculopatía) con déficit motor severo, mononeuritis múltiple asociada con crioglobulinemia, enfermedad desmielinizante con déficit motor, vasculitis de SNC con déficit focal, mielitis y meningoencefalitis como condiciones que ponen en riesgo la vida y requieren un manejo sistémico inmunosupresor111,112.
De lo anterior es preciso destacar que nuevamente se recomienda el uso de esteroides, a dosis de 0,5 mg a 1 mg /kg/día, pero se debe reducir lo más pronto posible y bajo la línea de la dosis más baja posible.
En la ganglionopatía y en la CIDP como complicaciones severas se han descrito ensayos con inmunoglobulina intravenosa (IGIV) a dosis de 0,2 a 0,4 g/kg/día113–115, glucocorticoides, ciclofosfamida80, plasmaféresis114, azatioprina116, interferón alfa117, D-penicilamina80 y rituximab75, pero estos no han demostrado evidencia fuerte a favor. Después de la IGIV, el micofenolato mofetil a dosis de 2 g/día es el tratamiento más efectivo que se haya reportado78. El consenso deja como primera línea el uso de IGIV, como segunda línea los pulsos de esteroides, y de rescate la ciclofosfamida80,111,118,119.
El tratamiento con terapia inmunosupresora en el contexto de la neuropatía sensitivomotora se reserva para síntomas rápidamente progresivos o severos, o cuadros clínicos que se acompañen de déficit motor significativo. Se ha utilizado principalmente IVIG a dosis de 0,4 g/kg/día por cinco días en cohortes pequeñas72,115. La terapia con rituximab ha demostrado resultados en aquellos pacientes en los cuales la neuropatía se relaciona con vasculitis o crioglobulinemia. De lo contrario no ha demostrado beneficio75,111.
En los casos de mononeuritis múltiple, en los que se descarte compromiso por crioglobulinemia, la primera línea es el uso de esteroides combinados con inmunosupresores orales (azatioprina, micofenolato, metotrexate) o rituximab. En el caso de asociación a crioglobulinemia, las opciones de ciclofosfamida y plasmaféresis las reservan como terapia de rescate a manifestaciones por crioglobulinemia que pongan en riesgo la vida111.
En el sistema nervioso central, las vasculitis, la NMO y la meningoencefalitis tienen como primera línea el uso de esteroides junto con ciclofosfamida como recomendación. En caso de considerarse rescate, las opciones pueden ser rituximab, plasmaféresis o eculizumab en el caso de la concomitancia con NMO AQP4 positivos, dada la evidencia reciente111,120.
Hay varias opciones de tratamiento en la presentación de EM asociada a SSp, según el perfil de la enfermedad, la edad de aparición, la severidad y la carga de la enfermedad, dentro de los que se encuentran inyectables como el interferón, orales como la teriflunomida, el dimetil fumarato, el fingolimod y la cladribina, y monoclonales como el natalizumab, el alemtuzumab y el ocrelizumab, entre otros. Sin embargo, estos tratamientos deben contar con la supervisión de un neurólogo, luego de descartarse los diagnósticos diferenciales, debido a la menor prevalencia de EM vs. NMO en el SSp. El pronóstico de la enfermedad dependerá principalmente de las secuelas que dejen las lesiones, según sea su localización121.
ConclusiónA pesar de ser una entidad relativamente benigna, el SSp puede llegar a implicar gran morbilidad y mortalidad cuando involucra manifestaciones neurológicas. Por otro lado, si bien la presencia de comorbilidades psiquiátricas no amenaza la vida del paciente, estas son altamente prevalentes, lo que confiere una mayor carga de enfermedad y predispone a mayor deterioro en la calidad de vida. A raíz de lo anterior, y de que el espectro clínico de las complicaciones neuropsiquiátricas es notoriamente variado, es preciso que el paciente con SSp sea abordado desde una perspectiva multidisciplinaria, tanto con el fin de enfocar el abordaje diagnóstico de estas, como de dirigir acertadamente su tratamiento.
Conflicto de interésLos autores declaran que no tienen ningún conflicto de intereses en el presente trabajo.
