



El síndrome de Sjögren es una entidad de origen reumático, con características autoinmune complejas, en la que se ven comprometidas principalmente las glándulas salivales y las lagrimales. Tiene 2formas de presentación, una primaria y otra secundaria, y en ambas se observa una afección de las glándulas exocrinas. El espectro clínico del síndrome de Sjögren es muy heterogéneo y se clasifica en manifestaciones glandulares y extraglandulares, no excluyentes entre sí. Se recomienda que se haga un control médico más estricto a todo paciente que curse con una inflamación parotídea, púrpura, hipergammaglobulinemia y anticuerpos anti-SSa, anti-SSb, puesto que presenta mayor riesgo de cursar con una presentación sistémica grave. Los estudios poblacionales que han intentado describir la incidencia y la prevalencia del síndrome de Sjögren en diferentes países son hasta cierto punto discordantes entre un registro y otro. El síndrome de Sjögren es más frecuente en mujeres, pero en hombres predomina más la afectación ocular; puede presentarse en todas las edades, principalmente entre la tercera y la quinta décadas de la vida; en niños es raro. Se considera además como una conectivopatía frecuente, en la cual los datos de la tasa de incidencia global y de prevalencia se encuentran subestimados.
Sjögren's syndrome is an entity of rheumatic origin, with complex autoimmune characteristics, in which the salivary and lacrimal glands are mainly compromised. It has 2forms of presentation, one primary and the other secondary, and in both forms there is evidence of exocrine glands involvement. The clinical spectrum of Sjögren's syndrome is very heterogeneous and is classified into glandular and extra-glandular manifestations, but not mutually exclusive. It is recommended that all patients with parotid inflammation, purpura, hypergammaglobulinaemia, anti-SSa, and anti-SSb should be seen to have a greater risk of presenting with a severe systemic presentation, and it is recommended to carry out a more strict medical control. Population studies that have attempted to describe the incidence and prevalence of Sjögren's syndrome in various countries throughout the world are to some extent discordant between one registry and another. Although Sjögren's syndrome is more common in women, ocular involvement predominates in men, and it can occur in all ages, mainly between the third and fifth decades of life. In children it is rare. It is also considered as a common connective tissue disease, where the data on the global incidence rate and prevalence are underestimated.
El síndrome de Sjögren (SS) es una enfermedad reumática de carácter autoinmune, compleja, que afecta principalmente a las glándulas salivales y lagrimales1-5. Los pacientes suelen presentar sequedad ocular y oral e inflamación glandular, que son puestas en evidencia mediante biopsia6,7. Además, con frecuencia se observa sequedad cutánea, nasal y vaginal8,9. La tríada clásica de la enfermedad que exhibe la gran mayoría de los pacientes con SS está constituida por dolor musculoesquelético, fatiga y sequedad10. Hasta un 30 a un 50% de los pacientes con SS puede mostrar enfermedad sistémica. Además, existe un mayor riesgo de desarrollo de linfoma no Hodgkin, que ocurre en un pequeño porcentaje de pacientes10.
El SS se presenta en una forma primaria, no asociada a otras enfermedades, y en una forma secundaria que complica otras condiciones reumáticas11-13. Las enfermedades más comunes asociadas con el SS secundario son la artritis reumatoide y el lupus eritematoso sistémico11.
Desde el punto de vista fisiopatológico, el principal mecanismo descrito es la infiltración linfocítica intensa de las glándulas exocrinas, así como la hiperactividad de los linfocitos B, que ocasionan inflamación, daño del tejido glandular y deterioro en su función14,15.
La incidencia global de SS se estima en aproximadamente 7 por 100.000 personas-año; sin embargo, las estimaciones sobre su incidencia y prevalencia varían ampliamente según los criterios de clasificación específicos, el diseño del estudio y la población examinada16. Las tasas de incidencia más altas de acuerdo con los estudios se informaron en Europa y Asia17. En un estudio realizado en Colombia por Fernández-Ávila et al.18, sobre la prevalencia y las características demográficas del SS, se identificaron 58.680 casos de esta enfermedad en el territorio nacional, según datos obtenidos de la base de datos del Sistema Integral de Información de la Protección Social. Los investigadores calcularon una prevalencia en mayores de 18 años del 0,12%, de los cuales el 82% eran mujeres, con una relación mujer:hombre de 4,6:1. Los departamentos con mayor número de casos eran Bogotá D.C. (24.885), Antioquia (9.040) y Valle del Cauca (5.277); sin embargo, los departamentos con mayor prevalencia fueron Caldas (0,42%), Bogotá D.C. (0,32%) y Antioquia (0,14%). Aunque el SS puede presentarse en todas las edades, la mayoría de los pacientes se ubica entre la tercera y la quinta décadas de la vida, con una prevalencia mayor entre los 65 y los 69 años18.
Al igual que muchas enfermedades médicas crónicas, la depresión y la ansiedad pueden acompañar al SS, afectando a la calidad de vida de los pacientes19,20. Esto se demostró por medio de la puntuación en la escala de intrusividad de la enfermedad, realizada por el Devin, en la cual el impacto negativo en la calidad de vida de los pacientes con SS es comparable con el de la esclerosis múltiple o la terapia de reemplazo renal21. Por lo anterior, es importante hacer un diagnóstico preciso del SS mediante una evaluación exhaustiva para acelerar la derivación a un médico especialista de forma oportuna22.
El objetivo de este escrito es hacer una descripción detallada de la epidemiología y las manifestaciones clínicas sistémicas asociadas al SS, mediante una revisión sistemática de la literatura científica, de tipo cualitativo, presentando la evidencia en forma descriptiva y sin análisis estadístico.
MetodologíaEstrategia de búsqueda de informaciónLa literatura para esta revisión sistemática se identificó mediante el planteamiento de las siguientes preguntas problema: ¿cuál es la epidemiología del SS?, ¿cuáles son las principales manifestaciones clínicas asociadas al síndrome de SS?
Se llevó a cabo una revisión sistemática, a partir de consultas en las bases de datos PubMed, Clinical Key, Scopus, the Cochrane Library, Nature, Science Direct y Google Académico, en busca de artículos publicados hasta el 31 de julio del 2020. Se utilizaron los siguientes criterios de búsqueda, diseñados a partir de términos incluidos en el tesauro DeC (http://www.decs.bvs.br), o de sus equivalentes en inglés incluidos en el tesauro MeSH (http://www.meshb.nlm.nih.gov): [Sjögren AND Manifestaciones clínicas] OR, [Sjögren AND Epidemiologia] OR, [Xerostomia AND Manifestaciones clínicas] OR, [Xerostomia AND Epidemiologia] OR, [Síndrome Seco AND Manifestaciones clínicas] OR, [Síndrome Seco AND Epidemiologia] OR, [Sequedad ocular AND Manifestaciones clínicas] OR, [Sequedad ocular AND Epidemiologia]. Se hicieron búsquedas adicionales de información en las listas de referencias bibliográficas de los artículos incluidos en el estudio, para evitar la pérdida de información relevante.
Criterios de inclusión y exclusiónSe incluyeron todos los artículos encontrados correspondientes a revisiones sistemáticas, revisiones narrativas o reportes de casos, que incluyeran revisión de la literatura, metaanálisis y ensayos controlados aleatorizados. Se excluyeron los artículos que duplicaban información contenida en estudios más extensos, cartas al editor y artículos que contenían información incompleta o aquellos a los cuales no fue posible el acceso al texto completo.
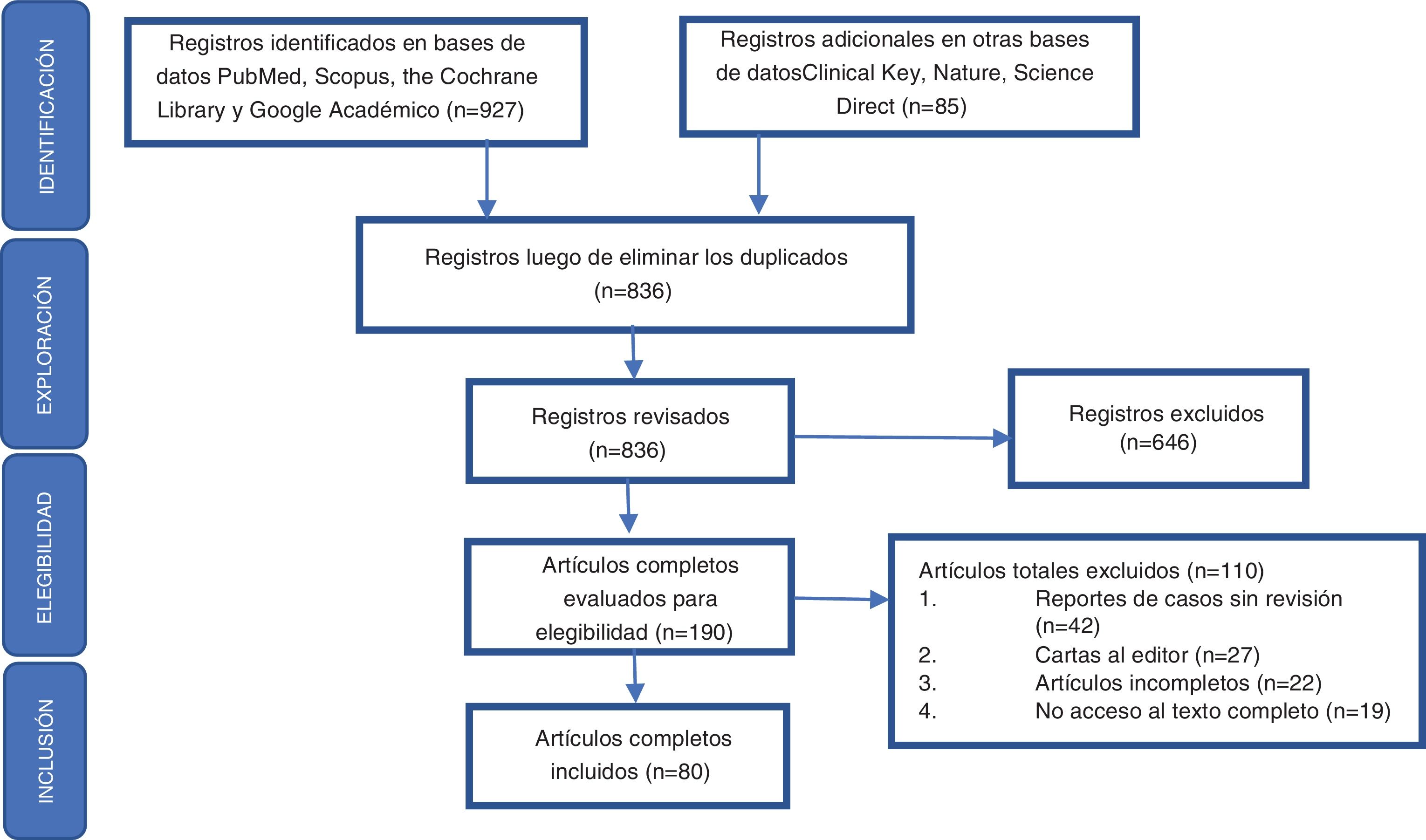
Extracción de datos y evaluación de la calidad de los artículos identificadosDe acuerdo con las preguntas problema planteadas, los autores se dividieron en 2grupos para llevar a cabo la identificación de la literatura, como resultado de lo cual se obtuvo un total de 836 registros en la búsqueda preliminar, para luego proceder de acuerdo con los elementos definidos en la declaración Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) a realizar la exploración, la elegibilidad y la inclusión de los registros obtenidos en la respectiva búsqueda bibliográfica (fig. 1). Después de la lectura crítica, fueron seleccionados 80 artículos y se ordenaron por temas aquellos considerados relevantes por los autores, debido a que incluían una revisión detallada de la epidemiología y las características clínicas glandulares y extraglandulares relacionados con SS. Se dio prioridad a aquellas publicaciones incluidas en revistas indexadas con un elevado factor de impacto de acuerdo con el Journal Citation Reports (JCR). Posteriormente se procedió al análisis y el desarrollo de la revisión.
Resultados y discusiónEpidemiología
El SS constituye una entidad de etiología autoinmune, de causa desconocida hasta el momento; es bien sabido no solo su marcado impacto en la calidad de vida de los pacientes que la presentan, sino también la carga de enfermedad que significa para cuidadores y sistemas de salud a escala mundial23. Las opciones terapéuticas efectivas son escasas y con ello se propicia el aumento de dicha carga24. Son diversos los estudios poblacionales que han intentado describir la incidencia y la prevalencia de SS en diversos países; los datos son hasta cierto punto discordantes entre un registro y otro, y van a depender muchas veces de los criterios diagnósticos utilizados para clasificar a los pacientes25.
Comparativamente, las tasas de incidencia de los estudios que utilizaron los criterios de la clasificación europea de 1993 eran muy inferiores, contrastadas con aquellas de los estudios realizados según los criterios del consenso Europeo/Americano de 200217. Las tasas de incidencia global oscilan entre 3 y 11 casos por cada 100.000 pacientes, mientras que la prevalencia se sitúa alrededor del 0,01-0,72%26,27. Muy probablemente estas cifras se encuentren infraestimadas, debido a que muchos pacientes asintomáticos nunca llegarán a ser diagnosticados28. En cuanto a la afectación por sexos, es mucho más frecuente en las mujeres, con una relación cercana de 10:1 (mujer:hombre), que fue reportada en un estudio que incluyo a más de 14.000 pacientes29. Sin embargo, hay otras diferencias en su comportamiento con respecto al sexo: en los hombres predomina la afectación ocular severa, mientras que las manifestaciones y el compromiso sistémico es mucho mayor en las mujeres30,31.
Aunque puede presentarse en todas las edades, la mayoría de los pacientes se ubican entre la tercera y la quinta décadas de la vida. En niños es mucho más rara, conservando la predominancia en mujeres, aunque con una menor proporción, con una edad promedio de inicio de la enfermedad entre 9,4 y 10,7 años, siendo las niñas menores a la edad del diagnóstico. A diferencia de los adultos, se ve una menor incidencia de síntomas de sequedad, pero mayor incidencia de parotiditis, linfadenopatía y síntomas sistémicos32,33. También se ha evaluado la posible injerencia de la raza o la etnicidad. En un estudio realizado en París (Francia) se documentó que la incidencia era 2veces mayor en población sin ascendencia europea que en aquella con ascendencia europea34. En poblaciones asiáticas se documentó una mayor prevalencia, en comparación con sujetos caucásicos26. De igual forma, se encontraron variaciones entre los estudios de un país y otro: es el caso de una prevalencia de 0,21% en Turquía, donde se usaron los criterios del Consenso Europeo/Americano; 0,77% en población china, usando los criterios de Copenhague, mientras que las tasas más bajas fueron reportadas en Japón, entre el 0,03 y el 0,05%35-38.
En Colombia, Santos et al.39 llevaron a cabo un estudio cuyo objetivo principal era describir la prevalencia de enfermedades reumáticas y factores asociados en la población colombiana mayor de 18 años, para lo cual incluyeron datos de 6ciudades principales y utilizaron el cuestionario Copcord (Programa orientado a la comunidad para el control de las enfermedades reumáticas) adaptado para Colombia. Posteriormente a este primer paso, los casos positivos eran evaluados en casa por un residente de primer año de Reumatología, en segunda instancia los evaluaba un residente de segundo año de Reumatología y, por último, con los resultados de imágenes y pruebas serológicas, fueron evaluados por un reumatólogo titulado. Los investigadores concluyeron que la prevalencia de SS era de 0,08%39. Estos datos son comparables a los publicados por Fernández-Ávila et al.18, quienes hicieron el análisis de poco más de 58.000 casos de SS, provenientes de la base de datos del registro oficial del Ministerio de Salud de Colombia, la mayoría de los pacientes procedentes de Bogotá, Antioquia y Valle del Cauca, y encontraron una prevalencia total del 0,12%, que varía ampliamente según el grupo etario evaluado, siendo inferior en los menores de 15 años y superior en la población entre 65 y 69 años (0,5%). La población femenina predominó, con una relación de 4,63:1 (mujer:hombre), lo que dio como resultado una prevalencia del 0,31% en mujeres y el 0,07% en hombres18.
Manifestaciones clínicasEl espectro clínico del SS es muy heterogéneo y se clasifica operatoriamente en manifestaciones glandulares y extraglandulares, las cuales no son excluyentes entre sí.
Manifestaciones glandulares exocrinasLa afectación de las glándulas exocrinas en pacientes con SS se manifiesta principalmente por hiposecreción, mediada por trastornos glandulares involucrados en la fisiopatología de la enfermedad. Estos síntomas son los más frecuentes y representan el punto clave para llegar al diagnóstico, aunque solo son referidos por los pacientes al momento de indagar por ellos40.
Enfermedad ocularLa xeroftalmia o queratoconjuntivitis seca, que es la manifestación ocular más frecuente en SS41, se presenta como sequedad y disminución del lagrimeo, prurito, sensación de cuerpo extraño, hiperemia de la conjuntiva y fotofobia42, lo cual genera alteraciones en la composición y el flujo lagrimal, rotura precoz de la capa lagrimal y lesiones en el epitelio ocular. Asimismo, aumenta la probabilidad de úlceras corneales, uveítis y escleritis43; en los casos moderados a severos se puede presentar neuritis óptica y queratitis filamentosa, entidad en la cual fibras proteínicas y de material mucoso se adhieren a la superficie corneal, lo que agrava los síntomas del ojo seco44.
La prevalencia de xeroftalmia alcanza al 15% de la población general, si bien de este porcentaje menos del 30% tiene SS, esta última entidad afecta a cerca del 90% de los pacientes diagnosticados con xeroftalmia44.
Enfermedad bucalA nivel bucal, la xerostomía es una manifestación prevalentemente descrita por los pacientes con SS45. La saliva es una secreción compleja, que proviene de las glándulas salivales mayores (parótida, sublinguales y submandibulares) en un 93% de su volumen y el 7% restante de las glándulas menores o secundarias (glándulas labiales, palatinas, genianas y linguales), que están distribuidas por toda la cavidad bucal46. Está compuesta de moléculas complejas como proteínas, glicoproteínas, lípidos, electrólitos, buffers, hormonas, inmunoglobulina A secretora, entre otras sustancias que desempeñan un papel importante en el mantenimiento de la salud bucal41. Por lo tanto, en pacientes con SS suelen presentarse complicaciones relacionadas con el déficit en la producción salival como caries, pérdidas dentales tempranas e infecciones orales recurrentes por Candida albicans. Estas condiciones son 10veces más frecuentes que en la población general y se manifiestan como lesiones en mucosa eritematosa, fisuras linguales, atrofia de las papilas filiformes y queilitis angular13,47,48.
También se ha reportado que en un 30-50% de los pacientes con SS existe hipertrofia o aumento de tamaño de las glándulas salivares, que puede iniciar de forma episódica o hacerse crónico. Este caso debe tener especial cuidado y vigilancia hasta excluir infecciones, y de forma más importante descartar linfoma con pruebas imagenológicas o de estudio patológico, ya que se calcula un riesgo del 4-10% de desarrollar un linfoma no Hodgkin a lo largo de la vida de un paciente con SS primario45.
Otras manifestaciones clínicas a nivel bucal informadas por pacientes con xerostomía (sicca oral) incluyen: halitosis, disgeusia, dificultad para hablar, dificultades para la adaptación de prótesis dentales y trastornos alimentarios secundarios49.
Otras afectaciones glandulares exocrinasDentro del espectro clínico del SS primario, se ha descrito además sequedad nasal en un 30% de los pacientes diagnosticados25, manifestada por costras nasales que provocan epistaxis recurrentes, así como alteraciones del olfato y de la mecánica respiratoria, lo que aumenta la xerostomía al respirar con la boca abierta al momento de dormir40.
La xerosis o piel seca es otra condición frecuente, encontrada en el 66% de los pacientes con SS. Aunque los síntomas de esta entidad son inespecíficos, se asocian con signos específicos como piel inelástica, áspera y descamativa, asociados con la hiposecreción de las glándulas sudoríparas50.
Por otro lado, no es extraño encontrar sequedad vaginal en la población femenina con SS, que se presenta con dispareunia y predisposición de infecciones vulvovaginales51. Las anteriores, si bien son situaciones multifactoriales, se deben tener en cuenta para el seguimiento de la enfermedad en estas pacientes.
Manifestaciones extraglandularesCualquier órgano o sistema puede verse afectado con intensidad variable por los fenómenos fisiopatológicos del SS52; en este apartado se describen las implicaciones clínicas del sistema extraglandular y de las glándulas no exocrinas. Estas manifestaciones pueden presentarse en diversas etapas de la enfermedad, por lo cual afectan su pronóstico y gravedad53.
Sistema musculoesqueléticoLa manifestación extraglandular más común en pacientes con SS es la artralgia y usualmente una poliartritis no erosiva que ocurre en aproximadamente el 50% de los casos, la cual puede aparecer antes de los síntomas secos en un 20%, simultáneamente en un 50% y después de estos en cerca de un 30% de los pacientes41,54. Una elevación de la prueba del factor reumatoide es característica de la artritis reumatoide y está presente en torno al 75% de los casos; sin embargo, también es positiva en un 60-70% de los pacientes con SS, y por esta razón no es de utilidad para distinguir una enfermedad de la otra41,55. Otras manifestaciones incluyen rigidez matutina, cuadros tipo fibromialgia, sin dejar de lado que un 70% de los pacientes presentan fatiga y afectación muscular en forma principalmente de mialgias. No obstante, se han descrito cuadros de debilidad muscular proximal de inicio insidioso o miopatía inflamatoria leve (polimiositis o miositis por cuerpos de inclusión)56.
Entre un 13 y un 65% de los pacientes muestran fenómeno de Raynaud, que puede preceder a los síntomas de sequedad y constituir por tanto el primer signo de la presencia de un SS primario, lo cual además se ha relacionado con la presencia de artritis, vasculitis, fibrosis pulmonar, glomerulonefritis, miositis y neuropatía periférica40,57.
Sistema hematológicoDentro de los trastornos hematológicos en pacientes con SS, es característica la anemia hasta en un 30-60% de los casos31,58. Usualmente, esta anemia es secundaria a la enfermedad inflamatoria crónica, no obstante, también se han documentado casos de anemia hemolítica, anemia perniciosa y anemia aplásica40,41,59. Otras alteraciones presentes incluyen la afectación de la serie blanca, que se manifiesta por leucocitopenia, linfopenia o eosinofilia58. Es frecuente además la presencia de hipergammaglobulinemia policlonal y crioglobulinas en suero, y en el caso de la crioglobulinemia mixta esencial debe descartarse la presencia de virus de la hepatitis C (VHC)40,60,61.
Sistema respiratorioEl compromiso respiratorio encontrado en SS va desde la afectación del tracto respiratorio superior hasta las pequeñas vías aéreas. Es importante el papel de la saliva como buffer del reflujo gástrico; su deficiencia, encontrada en los pacientes con SS, puede ocasionar tos y ronquera62.
Existen publicaciones sobre SS y desarrollo de enfermedad pulmonar intersticial, condición que afecta hasta el 25% de los pacientes63. Asintomática en la mayoría de los casos, también puede ocasionar disnea de manera progresiva, tos seca, dolor pleurítico e inclusive hipertensión pulmonar63,64. La enfermedad pulmonar intersticial en SS tiene varias formas de presentación, entre ellas, neumonitis intersticial específica, neumonitis intersticial linfocítica y neumonía criptogénica65. En pacientes asintomáticos se recomienda un seguimiento semestral o anual, en el que se realicen pruebas de imagen y de función respiratoria66.
Afectación cutáneaEl compromiso cutáneo, que se observa al menos en la mitad de los pacientes con diagnóstico de SS, se presenta con manifestaciones vasculíticas como púrpura hipergammaglobulinémica (15%), vasculitis leucocitoclástica (11%), urticaria vasculítica (21%), depósitos intraepidérmicos de IgG (66%), eritema anular y linfoma cutáneo de células B, cuya incidencia exacta aún no ha sido reportada50. La histología revela vasculitis leucocitoclástica en la dermis superficial, además de observarse ocasionalmente un infiltrado de células mononucleares que envuelve la pared vascular40. Dentro de las manifestaciones cutáneas no vasculíticas del SS se encuentra la xerosis, mencionada anteriormente en esta revisión, siendo la más frecuente, así como amiloidosis nodular, alopecia, anetodermia, vitíligo, síndrome de Sweet y liquen plano66-68.
Sistema gastrointestinalSe ha argumentado que síntomas gastrointestinales diversos que forman parte del SS y se ha propuesto que pueden obedecer a infiltración linfocítica de la mucosa gastrointestinal, o de las glándulas exocrinas, por neuropatías autonómicas o por el desarrollo de enfermedades autoinmunes asociadas41. Dentro de este espectro podemos encontrar disfagia, náusea, dispepsia, gastritis atrófica con infiltrados mononucleares de lámina propia y células T CD4 positivas y aclorhidria69. En los pacientes SS es obligatorio investigar la presencia de Helicobacter pylori, ya que este se ha asociado con linfoma de tejido linfoide asociado a mucosa (MALT)40,70.
A nivel hepático, el SS se relaciona con hepatomegalia y elevación de las enzimas hepáticas, hallazgo encontrado en un 10-40% de los pacientes71. Además, el diagnóstico de infección por VHC deberá excluirse del estudio del SS, ya que estos pacientes por lo general manifiestan sintomatología relacionada con el síndrome seco, aunque tienen una menor prevalencia de anticuerpos anti-Ro/La e hipertrofia parotídea y una mayor prevalencia de hipocomplementemia, crioglobulinemia y enfermedad hepática40,72. Otras condiciones intestinales descritas implican diarrea, enfermedad celiaca y disfunción pancreática41.
Sistema urinarioLa nefritis túbulo-intersticial y la acidosis tubular renal se han descrito como las formas más frecuentes de enfermedad renal en el SS, sin embargo, en la mayoría de los casos inician de forma sutil o de manera asintomática73. Es frecuente observar también casos de glomerulonefritis, por lo cual ante su sospecha debe excluirse la posibilidad de LES subyacente o crioglobulinemia74. Los síntomas a nivel vesical se pueden manifestar con disuria, polaquiuria, nicturia y urgencia; si no existe infección urinaria los síntomas pueden ser secundarios a cistitis intersticial; estos síntomas son 20 veces más frecuentes en las pacientes con SS que en la población general40,75.
Sistema nervioso central y periféricoLa prevalencia de la afectación neurológica en el SS es de aproximadamente el 20%, implicado con mayor frecuencia el sistema nervioso periférico (SNP) que el sistema nervioso central (SNC)41. Los síntomas neurológicos incluyen cefalea y disfunción cognitiva y afectiva, reportada esta última hasta en un 70% de los casos41,76. Habitualmente, la neuropatía precede al diagnóstico de SS hasta en un 81% de los pacientes77 y puede presentarse clínicamente como polineuropatía simétrica axonal sensitivo-motora. Esta, que es la afectación más frecuente del SNP, inicia con predominio de síntomas sensitivos o con neuropatía sensitiva pura o con neuropatía periférica «en guante y calcetín» en más del 10% de los pacientes40,54,78.
La polineuropatía simétrica axonal sensitivo-motora genera parestesias leves e indolentes de las extremidades inferiores distales y puede involucrar además las extremidades superiores en aproximadamente el 20% de los casos. En esta condición no se documenta denervación en los estudios electrofisiológicos79, con excepción de los casos de neuropatía dolorosa de fibras pequeñas, en los cuales la electromiografía puede mostrar afectación concomitante de fibras largas y una disminución de la densidad de las fibras nerviosas epidérmicas, o bien morfología anormal en biopsias cutáneas40,80. También se ha encontrado aparición de neuropatía atáxica sensitiva aguda, mononeuritis múltiple y neuropatía craneal múltiple76.
En el caso del SNC, se han comunicado manifestaciones como hemiparesia transitoria, neuritis óptica, convulsiones, ataxia, parkinsonismo, meningoencefalitis aséptica, diversas formas de mielopatía aguda y crónica, vasculitis, linfomas, alteraciones afectivas y demencia40,41.
Afectación de la glándula tiroidesAdemás de las implicaciones de las glándulas exocrinas documentadas en el SS, es también notable la clínica que produce a nivel de la función endocrina de la tiroides, siendo bien documentados los casos de tiroiditis autoinmune con anticuerpos contra la tiroglobulina, la antiperoxidasa y frente a las hormonas tiroideas, lo que ocurre en el 15% de los pacientes. Se han descrito también hipotiroidismo e hipertiroidismo no autoinmunes, así como hiperplasia tiroidea44,45.
La tabla 1 muestra un resumen de los estudios realizados en humanos relacionados con las manifestaciones clínicas glandulares y extraglandulares en SS.
Resumen de los estudios relacionados con las manifestaciones clínicas glandulares y extraglandulares en el síndrome de Sjögren
| Autor y año | Muestra | Método | Objetivo | Tipo de estudio | Resultado | Conclusión |
|---|---|---|---|---|---|---|
| Alani et al.1 | Estudios seleccionados=42 en totalLUES+SS=13 estudiosTotal de la muestra=8.166 pacientesTotal pacientes con SS=1.247 (15,2%)AR+SS=19 estudiosTotal de la muestra=6.626 pacientesTotal pacientes con SS=855 (12,9%)ES+SS=7 estudiosTotal de la muestra=1.108 pacientesTotal pacientes con SS=167 (15%)Miositis+SS=3 estudiosES+SS=7 estudiosTotal de la muestra=285 pacientesTotal pacientes con SS=25 (8,7%) | Búsqueda sistemática en las bases de datos PubMed y Embase hasta marzo del 2016 para identificar todos los datos publicados sobre tasas de prevalencia, perfil demográfico, manifestaciones clínicas, características de laboratorio y causas de muerte asociadas con SS. Las tasas de prevalencia de SS se resumieron con IC del 95% | Evaluar las tasas de PR y las características clínicas y serológicas del SS | Revisión sistemática de la literatura y metaanálisis | La PR combinada para el SS+AR fue del 19,5% (IC del 95%: 11,2-27,8), la PR para SS+LUES fue del 13,96% (IC del 95%: 8,88-19,04). La proporción mujer/hombre de SS en la población con AR fue 14,7 (IC del 95%: 7,09-256) y en la población con LUES fue de 16,82 (IC del 95%: 1,22-32,4) | Las tasas de prevalencia de SS varían ampliamente en diferentes poblaciones. Ambos metaanálisis realizados en las poblaciones con AR y LES se caracterizaron por un alto grado de heterogeneidad del estudio. Los resultados de este metaanálisis destacan la necesidad de estudios poblacionales de mejor calidad |
| Sacsaquispe et al.24 | Total, de la muestra=367 biopsiasEdad media=53 años (rango: 13-86 años), con predominio de mujeres (90,2%) | Estudio retrospectivo, en el que se evaluaron los formularios de solicitud de examen anatomopatológico y los portaobjetos histopatológicos. Los datos registrados incluyeron edad, sexo, características clínicas y grado de gravedad | Determinar la epidemiología, las características clínicas y el grado de gravedad del SS en biopsias de un laboratorio de patología oral | Estudio de cohorte retrospectivo | Después de analizar los casos se encontró que la xerostomía se manifestó en el 94,67%, mientras que la xeroftalmia en el 81,52% de los casos; la afectación sistémica predominantemente observada fue la artritis reumatoide (34,17%), además de la esclerosis sistémica y el lupus eritematoso. Los grados de gravedad más comunes entre los casos fueron el 2 (36,42%) y el 3 (33,9%). También se encontraron algunos folículos linfoides; es posible evaluar el progreso de la enfermedad y detectar precozmente el linfoma | El estudio muestra que el 43,98% de los casos se diagnosticaron más tarde en el curso de la enfermedad en los grados 3 y 4, lo que sugiere el diagnóstico en etapas avanzadas de la enfermedad |
| Virdee et al.32 | Población masculina con SS: 7 estudios incluidos. Total de la muestra=210 pacientesPoblaciones pediátricas con SS: 5 estudios incluidos. Total de la muestra=132 pacientes | Búsqueda de artículos en PubMed utilizando los términos MeSH: «síndrome de Sjögren», «hombres», «niño», «pediatría»; se extrajeron los datos concernientes a características epidemiológicas, clínicas y de laboratorio encontradas en las poblaciones masculina y pediátrica con SS primario, para analizarlos con herramientas de ayuda y posteriormente desglosarlos en tablas | Analizar las características epidemiológicas, clínicas y de laboratorio de los pacientes masculinos y pediátricos con SS primario | Estudio de corte transversal | El rango de la edad de inicio de la enfermedad fue de 9,4-10,7 años para los niños y de 39,4-56,9 años al diagnóstico para pacientes varones. Se identificó una prevalencia de manifestaciones extraglandulares entre el 52,6 y el 92,3% en la población masculina y del 50,0 al 84,6% en los niños, mientras que la sialometría anormal solo se reportó en la población pediátrica, con una prevalencia entre 71,4 y 81,8%. Variación significativa de marcadores serológicos positivos, con anticuerpos anti-Ro reportados entre el 15,7 y el 75,0%, y entre el 36,4% y el 84,6%, y anticuerpos anti-La entre el 5,6 y el 51,7%, y el 27,3 y el 65,4%, en la población masculina y pediátrica, respectivamente. Las características del SSp en la población masculina y en la pediátrica variaron según los diferentes estudios. En comparación con datos disponibles de poblaciones adultas con SSp, en niños diagnosticados con SSp se informó menos sequedad y mayor prevalencia de parotiditis, linfadenopatía y síntomas sistémicos y masculinos; los pacientes eran más jóvenes en el momento del diagnóstico. | La revisión contribuye a una mejor comprensión de la epidemiología del SSp en poblaciones rarasSe necesitan estudios grandes de cohorte longitudinal que comparen pacientes masculinos con femeninos y adultos con pacientes pediátricos |
| Zaldívar Pupo et al.48 | 150 artículos a texto completo, arbitrados, escritos en inglés y español, principalmente desde el año 2011 hasta junio del 2017 | Se realizó una revisión bibliográfica exhaustiva en las principales bases de datos de Infomed (SciELO Regional, Clinical Key, PubMed) del 12 enero al 25 de junio del 2017No se pusieron restricciones a los tipos de artículos revisados, aunque se priorizaron los artículos originales y las revisiones bibliográficas | Actualizar los signos y los síntomas orales, así como el manejo estomatológico, en pacientes con SS | Revisión sistemática de la literatura | Los principales síntomas orales del SS son: ardor y dolor de origen mucoso, disgeusia, dificultad en la fonación, formación del bolo alimenticio, masticación y deglución. Entre los signos orales se encuentran: pérdida del brillo, palidez y adelgazamiento de las mucosas, inflamación y candidiasis oral. El tratamiento estomatológico se realiza en 3fases: inicial, paliativa y preventiva, restauradora y rehabilitadora, y mantenimiento. | Es importante conocer y diagnosticar los signos y síntomas orales del paciente con SS, pues estos requieren un manejo estomatológico especial |
| Yayla et al.52 | Total de la muestra=352 pacientes con SSp | Estudio retrospectivo de los datos de los pacientes a los cuales subdividieron en 2 grupos:edad de inicio 35 años o menos (inicio temprano) y edad de inicio mayor de 35 añosSe compararon las características clínicas, de laboratorio y serológicas de los 2grupos, p <0,05 se consideró estadísticamente significativo | Investigar las características clínicas y de laboratorio de los pacientes con SS primario de inicio temprano | Estudio observacional retrospectivo | Se analizaron 40 pacientes del grupo con una edad de inicio de 35 o menos (11,4%) y 312 pacientes con una edad de inicio de más de 35 (88,6%). La frecuencia de afectación cutánea (22,5% vs. 1,9%, p <0,001) y renal (10% vs. 2,2%, p=0,026) fue significativamente mayor en el grupo de inicio temprano que en el grupo de inicio tardío. No hubo diferencias significativas entre los 2grupos en términos de xerostomía, sequedad ocular, artritis y otras afecciones sistémicas. La positividad anti-Ro52 (p=0,04), los niveles elevados de IgG en suero (p=0,004) y la presencia baja de C4 (p=0,002) fueron más frecuentes en el grupo de inicio temprano. | Se observó que el fenotipo clínico de los pacientes con SSp de inicio temprano puede ser diferente del de aquellos con inicio tardío. La observación más frecuente de factores de mal pronóstico en edades de inicio temprano, especialmente, muestra la necesidad de monitorear a estos pacientes con mayor regularidad |
| Payet et al.55 | Total de la muestra=294 pacientes16 pacientes con ACPA positivos con SSp278 pacientes ACPA negativos | En este estudio se incluyeron pacientes con SSp positivos y negativos para ACPA. Para los pacientes con ACPA positivo se realizó una reevaluación clínica y radiológica sistemáticamente después de al menos 5 años de seguimiento. El diagnóstico se revaluó al final del seguimiento para identificar a los pacientes que desarrollaron AR según los criterios de clasificación del American College of Rheumatology 1987. | Comparar a pacientes con ACPA positivos y negativos con SSp y evaluar el riesgo de evolución a AR | Estudio de cohorte | Los pacientes con ACPA positivos tenían con mayor frecuencia artritis (43,7% vs. 12,2%; p=0,003), pero no artralgias. También tenían afectación pulmonar más frecuente (25% vs. 8,1%; p=0,05). Después de una mediana de seguimiento de 8 (5-10) años, 7/16 (43,8%) pacientes desarrollaron AR, incluidos 5 (31,25%) con erosiones típicas de la AR. La elevación de los reactantes de fase aguda en la inclusión fue el único parámetro asociado con la progresión a AR erosiva | El seguimiento a término medio de los pacientes con SSp con ACPA positivos mostró que casi la mitad de ellos desarrolló AR, particularmente en presencia de elevación de los reactantes de fase aguda. Estos resultados apoyan la utilidad de una estrecha monitorización radiológica de estos pacientes para la detección temprana de cambios erosivos y no retrasar el inicio de un tratamiento eficaz. De hecho, algunos de estos pacientes con SSp positivo para ACPA pueden tener AR y SS asociados |
| Malladi et al.58 | Total de la muestra=1.927 participantes inscritos en el registro SICCA | Se seleccionó a 886 participantes que cumplieron con los criterios del AECG de 2002 para SSp, 830 casos «intermedios» que tuvieron algunos hallazgos objetivos de SSp pero no cumplieron con los criterios de AECG, y 211 individuos de control. Se estudió la prevalencia de anomalías inmunológicas y hematológicas de laboratorio; hallazgos específicos del examen reumatológico, y el médico confirmó enfermedad de tiroides, hígado, riñón y linfoma entre los participantes del SICCA. | Estudiar la prevalencia de manifestaciones extraglandulares en el SSp entre los participantes inscritos en el SICCA | Estudio de corte transversal | Las anomalías de laboratorio, incluidas las hematológicas, la hipergammaglobulinemia y la hipocomplementemia, ocurrieron con frecuencia entre los casos de SSp y fueron más frecuentes entre los casos intermedios que entre los participantes de control. La vasculitis cutánea y la linfadenopatía también fueron más frecuentes entre los casos de SSp. En contraste, la frecuencia de diagnósticos confirmados por el médico de enfermedad tiroidea, hepática y renal, y linfoma fue baja, y solo la cirrosis biliar primaria se asoció con el estado de los casos de SSp. Los síntomas reumatológicos y neurológicos fueron comunes entre todos los participantes del SICCA, con independencia del estado del caso | Los datos del registro internacional SICCA apoyan la naturaleza sistémica del SSp, que se manifiesta principalmente en términos de anomalías inmunológicas y hematológicas específicas. La aparición de otros trastornos sistémicos en esta cohorte es relativamente poco común. Las asociaciones informadas previamente pueden ser más específicas para seleccionar subgrupos de pacientes, como los referidos para la evaluación de ciertas manifestaciones neurológicas, reumatológicas o sistémicas |
| Abbara et al.64 | Total de la muestra=467 pacientes, de estos 21 con anti-RNP positivo y 446 con anti-RNP negativo | Se seleccionó a pacientes que cumplían los criterios del ACR/EULAR 2016 para SSp y tenían anticuerpos anti-RNP, sin otras enfermedades del tejido conectivo diagnosticadas y sin anticuerpos anti-dsDNA, recuperados de la base de datos del Centro Nacional de Referencia de Francia; se compararon con todos los demás pacientes con SSp con anticuerpos anti-Sm, anti-RNP y anti-dsDNA negativos | Describir y comparar las características clínicas y biológicas de sujetos con síndrome de SSp con y sin anticuerpos anti-RNP | Estudio de casos y controles | Los pacientes anti-RNP positivos tenían una mediana de edad más baja al inicio de los síntomas de SSp (41,0 vs. 50,0 años, p=0,01), una mediana de índice de actividad de la enfermedad del SS EULAR más alta en el momento de la inclusión (8,0 vs. 3,0, p <0,01), con mayor frecuencia síntomas constitucionales (14,3% vs. 0,01%, p <0,01), miositis (19,0% vs. 2,3%, p <0,01) y afectación pulmonar (19,0% vs. 5,7%, p=0,04). Además, los pacientes anti-RNP positivos tenían niveles medios más altos de gammaglobulina (22,5 frente a 13 g/l, p <0,01), con mayor frecuencia anticuerpos anti-SSA (90,5% vs. 67,1%, p=0,03), pero menos frecuente sialadenitis linfocítica con una puntuación de enfoque ≥ 1 (66,7% vs. 85,5%, p=0,03). Si el análisis se limita a pacientes positivos para anti-SSA, la positividad para anti-RNP se asocia con las mismas características clínico-biológicas, excepto la afectación pulmonar. | Los pacientes con SSp con anticuerpos anti-RNP mostraron una enfermedad sistémica más activa, con afectación muscular y pulmonar más frecuente, y aumento del nivel de gammaglobulina, en comparación con los pacientes anti-RNP negativos |
| Villon et al.68 | Total de la muestra=596 pacientes de 3cohortes | Se utilizaron 2 cohortes francesas en la cuales se evaluó la prevalencia de trastornos cutáneos en 395 pacientes con SSp, y diapSS. Fueron examinados 91 pacientes consecutivos con SSp por un dermatólogo y datos basales del ensayo aleatorizado TEARS (110 pacientes con diagnóstico reciente o SSp activo, tratados con rituximab o placebo, y evaluado para la sequedad de la piel usando una escala analógica visual de 100) | Determinar la prevalencia y la importancia de los trastornos dermatológicos en el SSp | Estudio de corte transversal | Las manifestaciones cutáneas incluidas en el ESSDAI fueron raras en la cohorte ASSESS (n=16/395, 4,1%, principalmente púrpuras; solo 3 tenían actividad alta) pero asociadas con actividad en los otros dominios ESSDAI (neurológico periférico (p <0,001), muscular (p=0,01), hematológicos (p=0,017) y biológicos (p=0,017)), antecedentes de artritis (p=0,008), esplenomegalia (p=0,024) y mayor nivel de gammaglobulinas (p=0,008). En comparación con los pacientes con SSp que no recibieron una consulta dermatológica, los pacientes con SSp que tuvieron una consulta dermatológica tuvieron significativamente más compromiso dermatológico fuera de la puntuación ESSDAI (42% [29/69] vs. 19,6% [11/56]; p=0,008). El estudio TEARS mostró una alta prevalencia de sequedad cutánea (EVA >50; 48,2%) y que estos pacientes con piel seca tenían mayor EVA de dolor (61,5±28,2 vs. 46,8±27,0; p=0,003) y sequía (79,4±15,2 frente a 62,5±21,7; p <0,0001). | El trastorno cutáneo más común es la sequedad, que se asocia con un mayor nivel de dolor y sequedad subjetiva general. La actividad cutánea de ESSDAI es rara, asociada con hipergammaglobulinemia y actividad de ESSDAI. El examen dermatológico sistemático es informativo para las lesiones de SSp no específicas |
| Guimarães et al.72 | Se describe el caso de una paciente femenina de 48 años con infección crónica por VHC y diagnóstico de lupus discoide con características clínicas de SS | Descripción del caso clínico y revisión de la literatura relacionada con lo descrito | Ilustrar la manera en que la presencia de infección crónica por VHC dificulta el diagnóstico de SS | Reporte de caso | El paciente presentó cuadro seco con hipocomplementemia y positividad a FR, características comunes en el VHC+SS, mientras que la crioglobulinemia estuvo ausente. No obstante, cumplió con los criterios de clasificación SSp (anticuerpo anti-SSA positivo, puntuación de tinción ocular superior a 5 y PCR negativa para ARN-VHC). Si bien la presencia de artritis y lupus discoide apoya una enfermedad reumática primaria, los parámetros inflamatorios normales, ANA negativos y biopsia salival son menos frecuentes en el SSp. | En conclusión, los pacientes infectados por el VHC pueden presentar síntomas de sequedad, y diferenciar las 2entidades, VHC+SS y SSp, puede resultar difícil. Se describe un escenario clínico en el que un paciente infectado por el VHC con características mixtas entre SSp y SS+VHC cumplió con los criterios de clasificación SSp ACR-EULAR 2016, y el ARN del VHC es imposible de rastrear mediante la técnica de PCR. |
| Luo et al.73 | En un estudio transversal se incluyó a 434 pacientes con SSp del Departamento de Reumatología del Primer Hospital afiliado de la Universidad Médica de Wenzhou de 2013 a 2017 | Se comparó a los pacientes con afectación renal con sus controles pareados por edad y sexo (SSp sin afectación renal). Se analizaron sistemáticamente las características demográficas, clínicas, histológicas, nefríticas e inmunológicas de la afectación renal en el SSp. Los posibles factores relacionados con la afectación renal se identificaron mediante análisis de regresión logística multivariante | Investigar las distintas características de la afectación renal en pacientes con SSp e identificar factores potenciales asociados con la afectación renal | Estudio transversal | 192 pacientes con SSp (88,48%) con afectación renal eran mujeres con una edad media de casi 58 años y una duración media de la enfermedad superior a 4 años. Se presentaron las manifestaciones clínicas, serológicas e inmunológicas y la clase de biopsia renal de los pacientes con SSp con afectación renal. Mediante análisis multivariados, la xeroftalmia, la positividad histológica para la biopsia de glándulas salivales inferiores (LSBG), los niveles de complemento 3 (C3) reducido anti-SSA/Ro52, la hipoalbuminemia y la anemia mantuvieron una asociación significativa con la afectación renal en SSp (todos p <0,05) | Además del patrón de LSGB, la positividad anti-SSA/Ro52, los niveles reducidos de C3, la hipoalbuminemia y la anemia también indican una asociación significativa con la afectación renal en el SSp. Por lo tanto, se requiere una vigilancia temprana para los pacientes con estas manifestaciones clínicas |
| Delalande et al.77 | Total, de la muestra=82 pacientes (65 mujeres y 17 hombres) con manifestaciones neurológicas asociadas con SSp | Se estudió retrospectivamente a 82 pacientes consecutivos con SSp remitidos a los departamentos de Neurología o Medicina Interna del Hospital Universitario de Lille entre enero de 1993 y diciembre del 2001. Todos los pacientes cumplieron con criterios americano-europeos para SS y tenían manifestaciones neurológicas durante el seguimiento | Describir la diversidad de complicaciones neurológicas derivadas del SS | Estudio observacional retrospectivo | La edad media de inicio del deterioro neurológico fue de 53 años. Esta afectación neurológica con frecuencia precedió al diagnóstico de SS (81%); 56 pacientes tuvieron trastornos del SNC, que eran en su mayoría focales o multifocales; 29 de los pacientes tenían compromiso de la médula espinal (mielopatía aguda [n=12], mielopatía crónica [n=16] o enfermedad de la neurona motora [n=1]); 30 pacientes tenían compromiso cerebral y 13 pacientes tenían neuropatía óptica. La enfermedad imitaba múltiples enfermedades remitentes recurrentes, como EM, en 10 pacientes, y EM primaria progresiva, en 13 pacientes. También se registraron síntomas difusos del SNC: algunos de los pacientes presentaron convulsiones (n=7), disfunción cognitiva (n=9) y encefalopatía (n=2); 51 pacientes tenían afectación del SNP. Sensoriomotor axonal simétrico,polineuropatía con predominio de síntomas sensoriales o puros, la neuropatía sensorial ocurrió con mayor frecuencia (n=28), seguida de compromiso del nervio craneal que afecta al trigémino, el facial o el coclear (n=16). Mononeuropatía múltiple (n=7), miositis (n=2),y también se observó polirradiculoneuropatía (n=1). | El estudio subraya la diversidad de afectación neurológica del SS. La frecuencia de manifestaciones neurológicas que revelan SS y el alto nivel de negatividad de los marcadores biológicos, especialmente en pacientes con afectación del SNC podrían explicar el elevado número de casos mal diagnosticados |
ACPA: anticuerpos antiproteína citrulinada anticíclica; ACR/EULAR: American College of Rheumatology/European League Against Rheumatism; AECG: Consenso americano-europeo; ANA: anticuerpos antinucleares; AR: artritis reumatoide; EM: esclerosis múltiple; ES: esclerosis sistémica; ESSDAI: EULAR Sjögren's Syndrome Disease Activity Index; FR: factor reumatoide; IC: intervalo de confianza; LUES: lupus eritematoso sistémico; PCR: reacción cadena de polimerasa; PR: prevalencia; SICCA: Registro de la Alianza Clínica Colaborativa Internacional de Sjögren; SNC: sistema nervioso central; SNP: sistema nervioso periférico; SS síndrome de Sjögren; SSp: síndrome de Sjögren primario; VHC: virus de la hepatitis C; LSBG: biopsia de glándula salival inferior.
El SS se considera una conectivopatía frecuente en la cual los datos de la tasa de incidencia global y de prevalencia se encuentran subestimados, y claramente es mucho más frecuente en mujeres. Se evidencia afección especial de las glándulas salivales y lagrimales, pero como se observó aquí existen afectaciones extraglandulares y su sintomatología puede variar de leve a severa. Además, se considera un modelo de enfermedad autoinmunitaria, en la cual se puede presentar un riesgo elevado de linfomas. Tiene una forma de presentación de inicio precoz, antes de los 35 años. Se recomienda que todo paciente que curse con una inflamación parotídea, púrpura hipergammaglobulinemia y anticuerpos anti-SSa y anti-SSb, que presenta mayor riesgo de cursar con una presentación sistémica grave, se haga un control médico más estricto. Por ello, es de mucha importancia el papel del clínico para detectar de manera exhaustiva los síntomas, los hallazgos clínicos y los resultados de laboratorios, evitando de este modo un sinnúmero de complicaciones en los pacientes.
FinanciaciónNinguna.
Conflicto de interesesNinguno.
