





El síndrome de Schnitzler es una enfermedad rara. En Colombia se la considera dentro de las enfermedades huérfanas, de carácter autoinflamatorio, y se la clasifica como tipo inflamosopatía adquirida compleja que produce clásicamente la presencia de rash urticarial, fiebre de larga duración, adenomegalias y artralgias que coexisten con pico gamma monoclonal típicamente de tipo IgM. Se presenta el caso de una mujer joven, con un cuadro larvado que inició con rash urticarial y características clínicas compatibles con el síndrome, y evidencia de pico monoclonal en electroforesis de proteínas que cumple con los criterios de Lipsker-Baltimore 2001, así como con los criterios de Estrasburgo 2013 para el diagnóstico, que presentó mejoría clínica tras la instauración de tratamiento inmunomodulador.
Schnitzler syndrome is a rare disease, in Colombia it is considered an orphan disease, of an auto-inflammatory nature, classified as a complex acquired inflammatory type of disease, which classically produces urticarial rash, long-standing fever, adenomegalies, and arthralgias coexisting with monoclonal gamma peak typically of the IgM type. We present the case of a young woman, with a larval picture that started with urticarial rash, with clinical characteristics compatible with the syndrome and evidence of monoclonal peak in protein electrophoresis meeting Lipsker-Baltimore 2001 criteria and Strasbourg 2013 criteria for diagnosis.
El síndrome de Schnitzler es una enfermedad crónica, poco frecuente, categorizada como autoinflamatoria y clasificada dentro de este espectro como inflamosopatía adquirida compleja1–3. El primer caso se describió en 19721,4,5 y, a pesar de su ya distante descripción en la literatura, aún no se superan los 300 reportes de casos y permanece como una enfermedad desconocida en el devenir médico cotidiano5. Este tipo de enfermedad afecta con un ligero predominio a varones, con una edad de aparición promedio de 40 a 50 años, pero se ha evidenciado gran heterogeneidad por medio de reportes de casos de pacientes desde adolescentes hasta octogenarios1,5,6. Se caracteriza clínicamente por la aparición de rash urticarial recurrente no prurítico, asociado con pico gamma monoclonal que puede asociarse a su vez con fiebre crónica, adenopatías, hepato u esplenomegalia y artralgias con presencia de dolores óseos6,7.
En el año 2001 se establecieron los criterios diagnósticos de Lipsker-Baltimore, que fueron reemplazados en 2013 por los criterios de Estrasburgo. Estos últimos han demostrado ser más prácticos en el ámbito clínico, ya que solo requieren la coexistencia de 2 criterios mayores (exantema urticarial crónico y gamapatía monoclonal IgM o IgG) y al menos 2 criterios menores (fiebre intermitente, artralgia o artritis, dolor óseo, linfadenopatía palpable, esplenomegalia o hepatomegalia, velocidad de sedimentación globular elevada, leucocitosis y anormalidades óseas)5.
La fisiopatología no ha sido aclarada totalmente y se ha tratado de comparar con otros síndromes autoinflamatorios, entre ellos el síndrome periódico asociado a criopiridinas, para dilucidar el mecanismo fisiopatogénico, el cual es compatible con la desregulación en la inmunidad innata mediada por neutrófilos y monocitos3,9,11,13.
No se ha podido determinar con claridad una base genética que explique la etiología de la enfermedad, pero se conoce que los pacientes presentan una secreción elevada de citoquinas tipo IL-1β e IL-6 por parte de las células monoclonales, lo que constituye un marcador de severidad de la enfermedad5.
A partir de lo anterior se ha correlacionado la elevación de citoquinas con la clínica de la enfermedad, atribuyéndose los efectos específicos de estas en distintos órganos, de tal manera que la IL-1B representa una molécula de señalización que induce un aumento de pirógenos endógenos en el sistema nervioso central (específicamente a nivel hipotalámico), con el subsecuente aumento de la temperatura corporal y la fiebre característica del síndrome9.
El aumento de la producción de IL-1β por parte de los mastocitos de la piel explica la presencia de urticaria crónica en los pacientes. El rol de la gammapatía monoclonal IgM aún no está dilucidado, ya que hay controversia sobre si este hallazgo es una causa o una consecuencia de la enfermedad. La hipótesis más acertada apunta a la estimulación persistente del receptor IL-1, lo cual pudiera conllevar una producción monoclonal de IgM1.
Desde esta perspectiva se ha constituido a la IL-1ß como diana farmacológica, y se han obtenido buenos resultados empleando el inhibidor anakinra, con niveles de remisión hasta del 83% de los pacientes1,9,11. El canakinumab también ha demostrado una adecuada efectividad en los pacientes1. El uso de otras medidas terapéuticas como corticoides, interferón alfa y colchicina tiene moderada efectividad y alto riesgo de reacciones adversas, por tanto, no se ha recomendado de forma universal1.
Se presenta el caso de una paciente joven con cuadro de urticaria crónica y manifestaciones clínicas posteriores que, con el sustento de los estudios de extensión, derivó en diagnóstico de síndrome de Schnitzler, una enfermedad rara de la que no se tiene documentación de casos en el ámbito local ni nacional. Por ende, constituye el primer reporte de caso de un paciente en el que se configura el diagnóstico de síndrome de Schnitzler en Colombia en función de los criterios de Estrasburgo.
Por lo señalado previamente, el conocimiento de esta enfermedad es relevante para ampliar el horizonte diagnóstico y establecer esta entidad nosológica dentro del espectro diferencial del paciente con confluencia de fiebre de origen desconocido y lesiones cutáneas, específicamente de tipo urticarial crónico. Basándonos en ello se pretende optimizar el conocimiento de una enfermedad infraestimada, con el fin de hacer un diagnóstico más temprano, el cual permita establecer un tratamiento oportuno que evite la exposición a medicamentos innecesarios y optimice la terapia precoz, de manera que lleve a un mejor pronóstico y disminuya la morbilidad y la mortalidad.
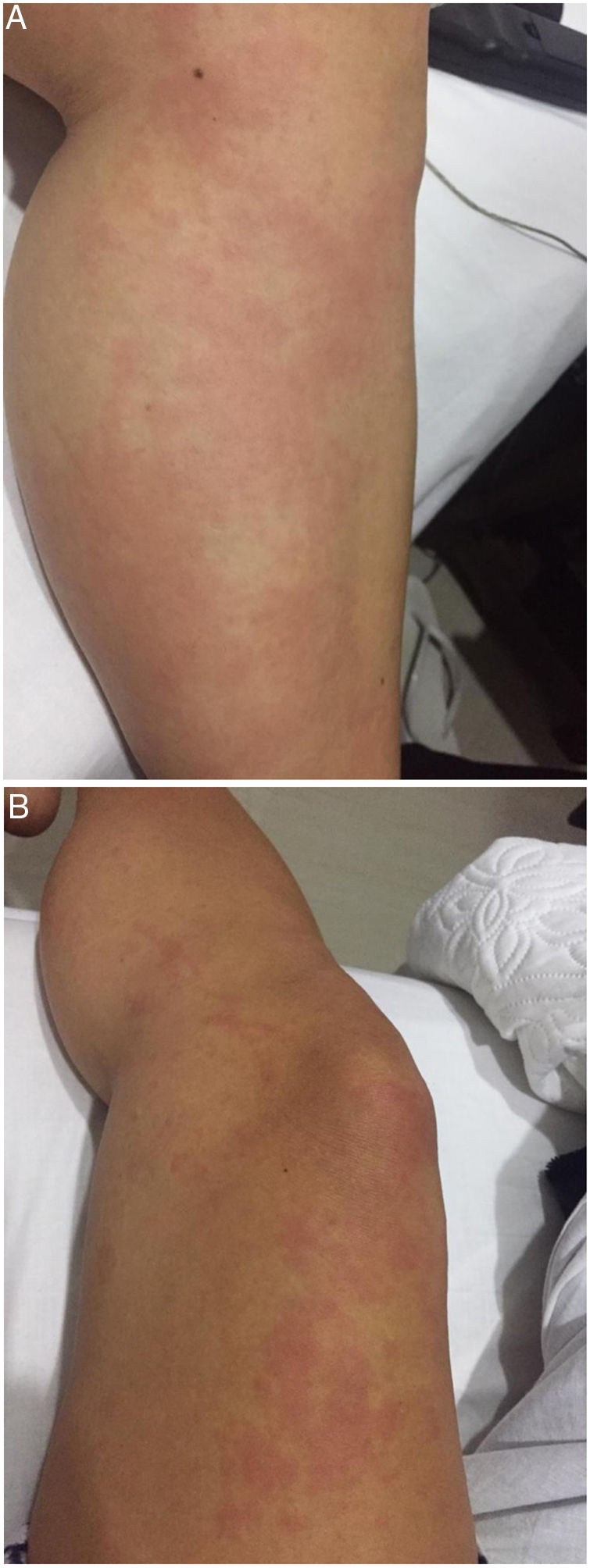
Presentación del casoPaciente mujer de 26 años, mestiza, con antecedente de tiroiditis autoinmune en seguimiento por endocrinología desde hace 6 años, quien presenta cuadro clínico de 4 meses de evolución que inició con rash urticarial en los miembros inferiores (fig. 1A y B), síntoma que solía iniciar en horas de la tarde y desparecer en la noche, sin evidencia de estigmas cutáneos en la mañana inmediatamente posterior. Con posterioridad, estas lesiones se diseminaron a los miembros superiores, el torso y la cara, con presencia concomitante de astenia y adinamia. Debido a esta situación clínica la paciente inicialmente consultó a dermatología y se le hizo diagnóstico de urticaria, a partir de lo cual se le instauró manejo médico con antihistamínicos, sin que experimentara mejoría alguna.

Tres meses después del inicio del cuadro clínico la paciente empezó a notar artralgias en las manos y las muñecas, que predominaban al despertar, acompañadas de rigidez matutina, sin cambios inflamatorios evidentes, pero con marcada mejoría en el curso del día. Luego se dio la aparición de ostealgias en la región anterior de la tibia, adenopatías en la cadena cervical y continuó con cuadro de escalofríos periódicos sin predominio horario alguno, lo que motivó una nueva consulta, esta vez por medicina interna y por reumatología. Como resultado de ello se solicitaron estudios de extensión y biopsia cutánea que reportaron cambios histopatológicos compatibles con urticaria, por lo cual se optimizó el manejo antihistamínico y la terapia en ciclo corto con glucocorticoide. Tras ello presentó mejoría parcial, pero con reanudación sintomática una vez retirado el medicamento.
Posteriormente a los síntomas descritos se superpuso la aparición de fiebre, que no tenía predominio horario, era fluctuante e inicialmente cedía a la administración de antipiréticos comunes. Finalmente, este cuadro febril se hizo continuo y llegó a presentar picos febriles hasta de 40°C, lo que motivó consultar por urgencias con la subsecuente hospitalización en unidad de tercer nivel de complejidad.
Al ingreso en la institución se describieron claros signos de respuesta inflamatoria sistémica, pero con qSOFA de 0 y apariencia general adecuada, sin signos de hipoperfusión tisular, con descripción de lesiones cutáneas jabonosas de distribución en el torso y las extremidades. Durante la estancia hospitalaria la paciente persistió con los síntomas y hubo recurrencia febril, a pesar de manejo con antipirexia y del establecimiento de manejo antibiótico múltiple de amplio espectro.
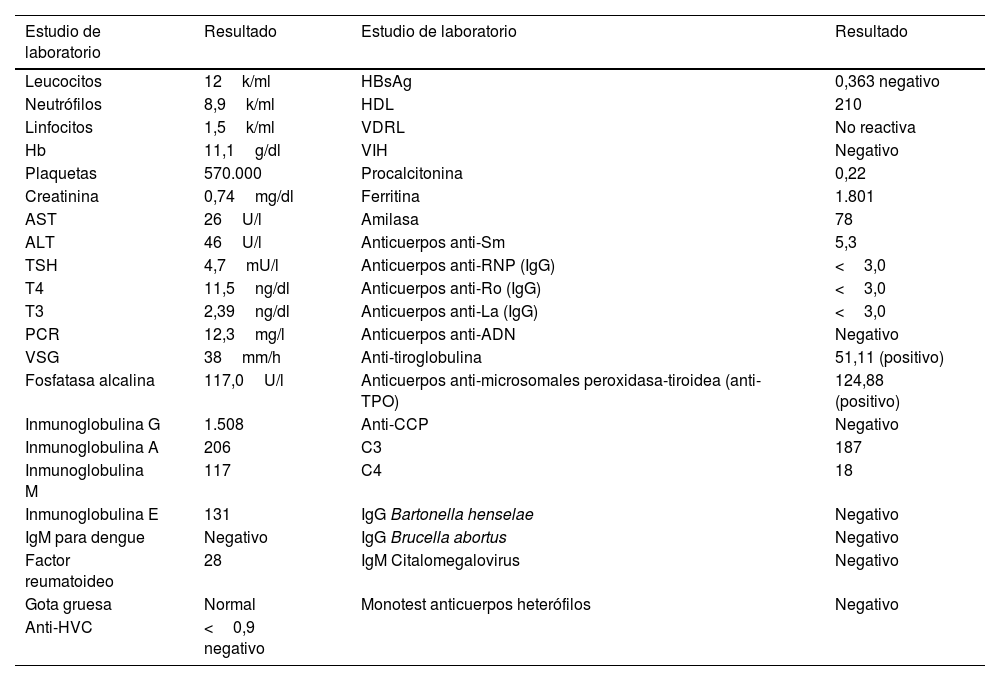
Durante la estancia hospitalaria se hicieron múltiples estudios de extensión que se representan en la tabla 1, así como estudios de imágenes en los que se documentó una tomografía simple (TAC) de tórax que mostró nódulos subpleurales de 3,2mm en el lóbulo medio y el superior derecho, con posible relación con granulomas en formación, además de una tomografía axial computarizada de abdomen y pelvis con hepatomegalia. En función de lo anterior se consideró un diagnóstico presuntivo de enfermedad de Still, pero a pesar de cumplir criterios mayores de Yamaguchi, como fiebre mayor de 39°C, artralgias, leucocitosis mayor de 10.000 y neutrofilia superior al 80%, más algunos criterios menores como linfadenopatías, había elementos en contra como la presencia de lesiones cutáneas, que no son las clásicamente descritas, como rash salmónico evanescente. De la misma manera hubo un resultado de factor reumatoide positivo con ferritina y pruebas de función hepática normales que hacían muy improbable esta hipótesis, máxime considerando que la enfermedad de Still supone un diagnóstico de exclusión. Con posterioridad se hizo un estudio de médula ósea que no mostró hallazgos patológicos y electroforesis de proteínas en suero y en orina, el cual encontró un pico monoclonal gamma e inmunofijación con pico monotípico IgG que en el contexto clínico de la paciente configuró el diagnóstico de síndrome de Schnitzler, cumpliendo criterios de Lipsker-Baltimore 2001, así como criterios de Estrasburgo 2013 (tablas 2 y 3). Una vez establecido el diagnóstico se inició un manejo con glucocorticoides en concomitancia con azatioprina, calcio y vitamina D, con lo que se dio la desaparición de las lesiones cutáneas, se produjo el cese de los picos febriles y la resolución de las artralgias y las ostealgias.
Estudios de paraclínicos realizados a la paciente
| Estudio de laboratorio | Resultado | Estudio de laboratorio | Resultado |
|---|---|---|---|
| Leucocitos | 12k/ml | HBsAg | 0,363 negativo |
| Neutrófilos | 8,9k/ml | HDL | 210 |
| Linfocitos | 1,5k/ml | VDRL | No reactiva |
| Hb | 11,1g/dl | VIH | Negativo |
| Plaquetas | 570.000 | Procalcitonina | 0,22 |
| Creatinina | 0,74mg/dl | Ferritina | 1.801 |
| AST | 26U/l | Amilasa | 78 |
| ALT | 46U/l | Anticuerpos anti-Sm | 5,3 |
| TSH | 4,7mU/l | Anticuerpos anti-RNP (IgG) | <3,0 |
| T4 | 11,5ng/dl | Anticuerpos anti-Ro (IgG) | <3,0 |
| T3 | 2,39ng/dl | Anticuerpos anti-La (IgG) | <3,0 |
| PCR | 12,3mg/l | Anticuerpos anti-ADN | Negativo |
| VSG | 38mm/h | Anti-tiroglobulina | 51,11 (positivo) |
| Fosfatasa alcalina | 117,0U/l | Anticuerpos anti-microsomales peroxidasa-tiroidea (anti-TPO) | 124,88 (positivo) |
| Inmunoglobulina G | 1.508 | Anti-CCP | Negativo |
| Inmunoglobulina A | 206 | C3 | 187 |
| Inmunoglobulina M | 117 | C4 | 18 |
| Inmunoglobulina E | 131 | IgG Bartonella henselae | Negativo |
| IgM para dengue | Negativo | IgG Brucella abortus | Negativo |
| Factor reumatoideo | 28 | IgM Citalomegalovirus | Negativo |
| Gota gruesa | Normal | Monotest anticuerpos heterófilos | Negativo |
| Anti-HVC | <0,9 negativo |
ALT: alanina aminotransferansa; AST: aspartato aminotransferansa; anti-CCP: anticuerpos antipéptidos cíclicos citrulinados; anti-HVC: anticuerpos contra el virus de la hepatitis; anti-TPO: anticuerpos anti peroxidasa tiroidea; HB: hemoglobina; HBsAg: antígeno de superficie de la hepatitis B, HDL: lactato deshidrogenasa; LDH: lactato deshidrogenasa; PCR: proteína C reactiva; VSG:velocidad de sedimentación globular.
Fuente: elaboración propia.
Criterios diagnósticos de Lipsker-Baltimore 2001
| Erupción, urticaria y componente monoclonal de IgM y al menos 2 de los siguientes criterios |
|---|
| Fiebre |
| Artralgia o artritis |
| Dolor de huesos |
| Ganglios linfáticos palpables |
| Agrandamiento del hígado o del bazo |
| VSG elevada |
| Leucocitosis |
| Hallazgos anormales en investigaciones morfológicas óseas |
Criterios diagnósticos de Estrasburgo
| Criterios obligatorios |
| Erupción urticaria crónica y IgM o IgG monoclonales |
| Criterios menores |
| Fiebre recurrentea |
| Hallazgos objetivos de remodelado óseo anormal con o sin dolorb |
| Infiltrado dérmico neutrofílico en biopsia de pielc |
| Leucocitosis o PCR elevadad |
| Diagnóstico definitivo si |
| Dos criterios obligatorios y al menos 2 criterios menores si es IgM y 3 criterios menores si es IgG |
| Diagnóstico probable si |
| Dos criterios obligatorios y al menos un criterio menor si es IgM y 2 criterios menores si es IgG |
Debe ser>38°C y sin otra explicación. Ocurre generalmente, pero no obligatoriamente, junto con la erupción cutánea.
Según lo evaluado por gammagrafía ósea, resonancia magnética o elevación de la fosfatasa alcalina ósea.
El síndrome de Schnitzler es una enfermedad autoinflamatoria crónica adquirida poco frecuente. La literatura ha reportado alrededor de 300 casos en el mundo, pocos de ellos informados en Latinoamérica5. La enfermedad se manifiesta con erupciones urticariales, fatiga, dolor óseo o articular, inflamación ganglionar y fiebre asociada con gammapatía monoclonal de tipo IgM; su fisiopatología aún no resulta clara1–3,5. La literatura ha reportado una edad promedio de 40 a 56 años, con predominio en hombres1,5,6. Nuestro caso, una paciente mujer joven de 26 años, es una variación a lo reportado.
Este caso representó un reto diagnóstico debido a los síntomas y a la posibilidad de un síndrome de Still, sin embargo, se hicieron hallazgos que mostraron que era poco probable que se tratara de esta enfermedad, como la presencia de lesiones cutáneas, que no son las clásicamente descritas, como rash salmónico evanescente. Adicionalmente, hubo un resultado del factor reumatoide positivo con ferritina y pruebas de función hepáticas normales, lo que hacía muy improbable esta hipótesis. Es de resaltar que fue indispensable la combinación de hallazgos clínicos, de laboratorio e imagenológicos. Se consideraron los criterios de Lipsker-Baltimore 2001, así como los de Estrasburgo 2013, además de la exclusión de otras causas como hepatitis autoinmune, pero no fueron concluyentes por pruebas de función hepática normales.
Como se mencionó con anterioridad, no se conoce muy claramente la patogenia de la enfermedad3,8-12; los diferentes estudios hacen referencia a una alteración del equilibrio de citoquinas de la inmunidad innata, por lo cual, al ser reconocida como un trastorno inflamatorio, las investigaciones actuales se han centrado en el papel de la IL-1β, de ahí que los fármacos inmunosupresores, como los corticoides, el interferón α, los inhibidores de la ciclooxigenasa, la anakinra, el antagonista del receptor de IL-1, la colchicina, la ciclosporina o la talidomida se utilicen para el control de la enfermedad1,5,9-11,13. Debido a su poca frecuencia aún no existen estudios que comparen la eficacia de los diferentes tratamientos, ni se ha reportado un manejo terapéutico estándar, por eso en nuestro caso utilizamos glucocorticoides en concomitancia con azatioprina, y obtuvimos buenos resultados y un control de los síntomas.
ConclusiónEl síndrome de Schnitzler es una enfermedad rara que no siempre se presenta en el rango de edad que habitualmente reporta la literatura; el diagnóstico es esencialmente de exclusión y es fundamental la combinación de hallazgos clínicos, de laboratorio y de imágenes, así como la colaboración multidisciplinaria. El manejo terapéutico sigue representando un desafío, y el uso de corticoides, así como de otros fármacos inmunosupresores, lleva al control óptimo de los síntomas.
Consideraciones éticasEl reporte de caso se realizó bajo el consentimiento informado de la familia, según la resolución n.o 8430 de 1993 del Ministerio de Salud de Colombia, garantizándose la confidencialidad de los datos del paciente según los principios de la Declaración de Helsinki. Los participantes en esta investigación fueron únicamente personas mayores de edad según la legislación colombiana, de quienes se obtuvo consentimiento informado por escrito para la recolección de información, el análisis de datos y la publicación de resultados, manteniéndose su anonimato durante todo el proceso.
Conflicto de interesesLos autores declaran que no existe ningún conflicto de intereses.
FinanciaciónNinguna.
