




Cutaneous mucinosis is a group of conditions characterized by the abnormal deposition of mucin in the skin. They can be primary, which in turn can be inflammatory-degenerative, and hamartomatous-neoplastic; or secondary. Papulonodular mucinosis is part of the group of dermal inflammatory-degenerative primary mucinosis. Its association with autoimmune connective tissue diseases has been described, especially with systemic lupus erythematosus, but it is considered an unusual manifestation of this disease.
The clinical case is presented of an 11 year-old girl who, at the onset of systemic lupus erythematosus, presented with skin lesions for which the histopathological diagnosis corresponded to mucinosis.
Las mucinosis cutáneas son un grupo de condiciones caracterizadas por el depósito anormal de mucina en la piel. Pueden ser primarias, que a su vez pueden ser inflamatorias-degenerativas (dérmicas o foliculares) y hamartomatosas-neoplásicas; o secundarias. La mucinosis papulonodular forma parte de las mucinosis primarias inflamatorias-degenerativas dérmicas. Se ha descrito su asociación con enfermedades autoinmunes del tejido conectivo, especialmente con el lupus eritematoso sistémico, pero se considera una manifestación inusual de esta enfermedad.
Se presenta el caso clínico de una niña de 11 años, quien al inicio del lupus eritematoso sistémico presentaba lesiones en la piel cuyo diagnóstico histopatológico correspondió a mucinosis.
Cutaneous mucinosis is a heterogeneous group of conditions characterized by abnormal focal or diffuse deposition of mucin in the skin. When mucin is deposited in the epidermis it is called sialomucin or epidermal mucin, and when it is deposited in the dermis or in the pilosebaceous follicles it is called dermal mucin and follicular mucin, respectively.1 They are classified into primary, where the mucin deposition is the main histopathological finding, producing clinically distinctive lesions; or secondary, in which the presence of mucin simply represents an associated finding. Primary mucinosis can be of 2 types: inflammatory-degenerative, which in turn are divided into dermal or follicular, and hamartomatous-neoplastic.2
Papulonodular mucinosis (PNM), also called papular and nodular mucinosis of Gold or lupus cutaneous mucinosis, is part of the group of dermal inflammatory-degenerative primary mucinosis, and is associated with autoimmune connective tissue diseases, most often with systemic lupus erythematosus (SLE), and to a lesser extent with dermatomyositis and scleroderma.2–4
The clinical case is presented of an 11-year-old patient with systemic, articular, cutaneous and laboratory manifestations typical of SLE, in whom was very striking the presence of papulo-nodular lesions in the skin of the proximal region of the extremities whose biopsy showed mucin, compatible with PNM
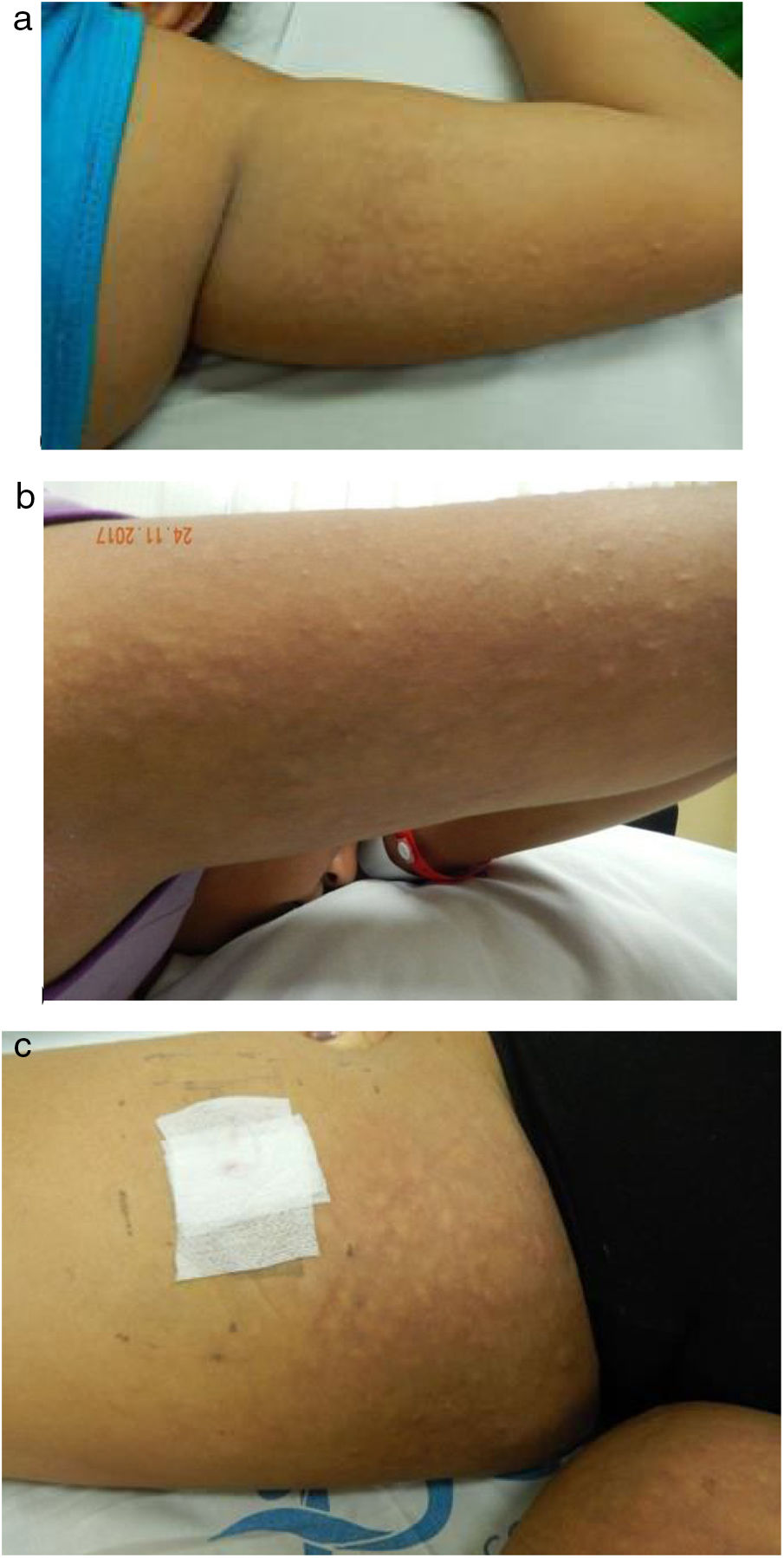
Clinical caseAn 11-years old female adolescent without relevant pathological antecedents, evaluated for a clinical picture of 20 days of evolution that started with polyarthralgias of inflammatory pattern, followed by cutaneous lesions of different morphology affecting the face, the back and the extremities, and subsequently onset of fever, asthenia, adynamia, hyporexia, hair loss, photosensitivity and malar erythema. On physical examination it was found a patient in a general fair condition, obese (body mass index of 25), who presented ulcers in the jugal mucosa and on the palate, multiple cervical adenopathies, hepatomegaly and polyarthritis of small, medium and large joints. In the skin, malar erythema and discoid lupus lesions were observed in the cheeks and in the pavilions of the ears. The most remarkable finding was the presence of multiple normochromic subcutaneous nodules with a cobblestone appearance, predominantly in the proximal region of arms and thighs. (Fig. 1). The laboratory tests reported mild thrombocytopenia (114,000/mm3 reference value [RV 150,000–450,000/mm3]), hypocomplementemia (C3: 23mg/dl [RV 82–173mg/dl], C4<2.9mg/dl [RV 13–46mg/dl]), positive antinuclear antibodies 1:1,280 homogeneous pattern (RV<1:40 negative), positive anti-native DNA 1:640 (RV negative), positive anti Ro (89.7 units [RV 0–20 units]), positive anti La (129.9 units [RV 0–20 units]), positive anti RNP (35.6 units [RV 0–20 units]), protein electrophoresis with polyclonal hypergammaglobulinemia (gamma fraction 25.8% [RV 6.9–14.7%]), normal total cholesterol (130mg/dl [RV 130–200]), low HDL cholesterol (17mg/dl [RV 40–60]) hypertriglyceridemia (triglycerides 515mg/dl [RV 7–150mg/dl]), elevated transaminases (AST 65IU/l [RV 5–45IU/l, ALT 65IU/l [RV 5–45IU/l) hepatosplenomegaly and hepatic steatosis documented by abdominal ultrasound.
 and in the thighs (c).")
Biopsies of the nodules of the left thigh and the back were taken, with the following report: “Back: skin with unchanged epidermis, in dermis there is a slight increase in inflammatory infiltrate, composed of lymphocytes and histiocytes, arranged in an interstitial pattern apparently associated with increased dermal mucin. Thigh: skin with unchanged epidermis and in the dermis and subcutaneous cellular tissue there is an increase in the interstitial mucin accompanied by scarce inflammatory infiltrate composed by lymphocytes, plasma cells and mast cells. Conclusion: presence of dermal and hypodermal mucin compatible with mucinosis associated with lupus” (Fig. 2a). The colloidal iron staining confirmed mucin deposits in the dermis and hypodermis. (Fig. 2b).
. The colloidal iron staining shows deposits of mucin in the dermis and the hypodermis (b, magnification 10×).")
Treatment was started with prednisolone 0.5mg/kg/day, azathioprine 1mg/kg/day and chloroquine 2.4mg/kg/day. There was resolution of the systemic and joint manifestations and a discrete improvement in the nodules was observed at 2 weeks, with complete resolution of the lesions at 4 months of follow-up.
DiscussionThe case reported illustrates a pediatric patient, who simultaneously with the onset of SLE presented cutaneous lesions with mucin deposits in the biopsy, compatible with PNM. In the Colombian literature this is the second case described,5 and the first in a patient with juvenile lupus.
Mucin is a component of the dermal extracellular matrix, which is produced in small quantities by fibroblasts and plays a primordial role in the hydrosaline balance of the dermis, given its ability to absorb 1000-fold its weight in water. It is composed of a mixture of acid glycosaminoglycans (acid mucopolysaccharides), which are complex carbohydrates that can be bound to proteins, as observed in proteoglycans such as heparan, chondroitin and dermatan sulfate, or be free as in hyaluronic acid.2,6
Although the histological finding of small mucin deposits is frequent in SLE lesions (secondary mucinosis), it is unusual for such a deposit to induce clinically detectable lesions such as papules or nodules.7 In 1954, Gold described for the first time this association when he reported 2 patients with SLE and discoid lupus erythematosus who had nodular deposits of mucin in the trunk, and highlighted that the deposits were not confined only to the areas of eruption of the lupus.8 Since the description of Gold, several cases have been reported, most of them as individual reports.6 Rongioletti described this manifestation in 1.5% of patients with SLE.9 In the pediatric population the association of SLE and PNM is unusual; we only found 2 cases reported.10,11 The first one in 1992, of a 13-year-old girl of Hispanic origin, with a history of 2 months of evolution of asymptomatic papular lesions in the trunk and lower limbs; the second published in 2007 of a 12-year-old girl, who consulted for purpuric lesions in the legs and arthritis in the ankles, and also presented multiple asymptomatic papulonodular lesions of 2–6mm in diameter in the trunk and upper limbs. Both cases, as well as the case presented in this report, had clinical and laboratory manifestations of active lupus. In the reports of adults, the relationship between the PNM and the disease activity is variable.12
The etiopathogenesis of cutaneous mucinosis is not well elucidated. The most accepted theory is the overproduction of glycosaminoglycans by dermal fibroblasts. The study conducted by Pandya et al.13 demonstrated that the serum of a patient with SLE and PNM induced a greater production of glycosaminoglycan by the fibroblasts compared with the serum of a healthy volunteer. These observations suggest that serum factors like immunoglobulins or cytokines such as IL-1, TNF-α and TGF-β, that positively regulate the synthesis of mucin, are present in patients with PNM. In addition, they observed that the papulonodular lesions increased concomitantly with the titers of circulating serum antibodies, suggesting that these antibodies could favor the synthesis of glycosaminoglycans. The reduction in the catabolic degradation of mucin, the exposure to ultraviolet radiation, the decrease in serum hyaluronidase and the vascular damage14,15 are other factors that may be implied.2
Clinically, the PNM is characterized by the presence of normochromic, or sometimes erythematous, papules and nodules which give the skin a “lumpy” appearance, and can sometimes come together forming plaques. They have a variable size between 0.5 and 2cm in diameter and are usually asymptomatic. Tangential lighting can allow a better visualization of the lesions. The most common locations are the back, the “V” of the chest and the upper limbs, but lesions may also appear on the face, the neck, the breasts and the lower limbs.2,15,16
For the histological diagnosis of all cutaneous mucinosis, 3 key factors should be considered: the distribution pattern of mucin, the affected level of the dermis and the presence of other distinctive histological findings.1 In the PNM there is an abundant and diffuse deposition of mucin between bundles of collagen in the upper dermis, although the mucin may also be occupying the subcutaneous cellular tissue. A scarce perivascular lymphocyte infiltrate may be observed, the direct immunofluorescence may reveal linear or granular deposits of IgG and IgM immunoglobulins and C3 at the dermoepidermal junction and epidermal changes of lupus are absent.2 In the cases reported in the Japanese literature, leukocytoclastic vasculitis has occasionally been seen.17 To confirm the presence of mucin, special colorations should be made with Alcian blue (it stains at a pH of 2.5), toluidine blue (it stains at a pH of 4) or colloidal iron.1
Among the main differential diagnoses of PNM are other cutaneous mucinosis such as lichen myxedematosus, myxedema, scleredema and reticular erythematous mucinosis; and other conditions such as lupus profundus and rheumatoid nodules.12,14 Myxedematous lichen is the main differential diagnosis; the papules are smaller and densely grouped, mainly in the face and arms; in the biopsy, the mucin deposit is located in the papillary dermis and frequently has large stellate fibroblasts.6
The treatment in general consists of the same therapy used for lupus, which includes topical, systemic and intralesional steroids and antimalarials.18 Approximately 20% of patients have a good response to antimalarials,19 and the treatment with systemic steroids has been successful in many patients, while in 13 case reports there was a significant improvement of the lesions with doses of prednisone greater than or equal to 60mg per daty.7,11,20 Although the reports are variable, in general, in most patients the lesions disappear after several months.20,21
In conclusion, the PNM is a rare and atypical manifestation of SLE, even more so in the pediatric population. In concordance with the previously reported cases, the diagnosis was made simultaneously with the SLE and it was associated with manifestations of activity.
Conflict of interestThe authors declare that there is no conflict of interest in the preparation and publication of this manuscript.
Thanks to the Hospital Pablo Tobón Uribe.
Please cite this article as: Ramírez Campo L, Londoño Salinas AM, Gómez Gómez LV, Ruíz Suárez AC, Eraso Garnica RM. Mucinosis papulonodular en el inicio de lupus eritematoso sistémico juvenil: asociación inusual. Rev Colomb Reumatol. 2019;26:282–286.
