




Antiphospholipid syndrome is frequently associated with systemic lupus erythematosus and other autoimmune diseases. However, coexistence with primary vasculitis has been poorly reported. The case is presented of a 67-year-old patient with a history of recurrent abortion and chronic pulmonary thromboembolism who was admitted due to haemoptysis. At the initial evaluation, a massive alveolar haemorrhage and glomerulonephritis were diagnosed. The results of the antibodies were positive for ANCA with P-type pattern, anti-myeloperoxidase antibodies, and antiphospholipid antibodies (anti-β2 IgG glycoprotein 1 and lupus anticoagulant). Diagnosis of ANCA positive vasculitis-type microscopic polyangiitis was made in association with antiphospholipid syndrome. Given the clinical context, it was decided to initiate intravenous methylprednisolone in pulses for 3 consecutive days, followed by oral prednisone, and as maintenance therapy, rituximab and anticoagulation with warfarin were instituted. The clinical evolution of the patient was satisfactory, with symptom control being achieved, as well as a significant improvement of renal and pulmonary function, with a decrease in the Birmingham vasculitis activity score (BVAS).
El síndrome antifosfolípido se asocia frecuentemente con lupus eritematoso sistémico y otras enfermedades autoinmunes. Sin embargo, la coexistencia con vasculitis primaria ha sido poco reportada. Se presenta el caso de una paciente de 67 años de edad con historia de aborto recurrente y tromboembolismo pulmonar crónico, quien es admitida para estudio de hemoptisis. A la evaluación inicial se diagnosticó una hemorragia alveolar masiva y glomerulonefritis. El resultado de los anticuerpos fue positivo para anticuerpos anticitoplasma de neutrófilos (ANCA) con patrón tipo perinuclear, anticuerpos anti-mieloperoxidasa y anticuerpos antifosfolípidos (anti β2 glicoproteína1 IgG y anticoagulante lúpico), configurándose el diagnóstico de vasculitis asociada a ANCA de tipo poliangitis microscópica en asociación con síndrome antifosfolípido. Dado el contexto clínico, se decidió iniciar metilprednisolona intravenosa en pulsos por 3días consecutivos, seguida de prednisona oral, y como terapia de mantenimiento se instauró rituximab y anticoagulación con warfarina. La evolución clínica de la paciente fue satisfactoria, alcanzando control de síntomas e importante mejoría de la función renal y pulmonar, con disminución del score BVAS.
Antiphospholipid syndrome (APS) is a condition defined by the coexistence of venous or arterial thrombosis, or gestational morbidity in the presence of antiphospholipid antibodies (lupus anticoagulant, anticardiolipin or anti β2 glycoprotein (1) persistently positive for more than 12 weeks.1 APS is frequently associated with systemic lupus erythematosus or with other underlying autoimmune disease, such as hemolytic anemia, idiopathic thrombocytopenic purpura, rheumatoid arthritis, Sjögren's syndrome, systemic sclerosis, mixed connective tissue disease, polymyositis, among others2; however, its coexistence with primary systemic vasculitis is infrequent.3
Microscopic polyangiitis (MPA) is a necrotizing pauci-immune vasculitis, which predominantly affects small vessels (e.g., capillaries, venules or arterioles).4 Its onset is more frequent in people over 50 years of age and affects more men than women; the two organs typically affected and that often define the prognosis are the kidney and the lung; however it can affect concomitantly other organs, such as the nervous system, the skin, the musculoskeletal system, the heart, the eyes and the gastrointestinal tract.5
A search conducted in Pubmed on the coexistence of MPA and APS only demonstrated one case published in this database.
The case is presented of a patient with a diagnosis of positive myeloperoxidase-anti-neutrophil cytoplasmic antibodies (MPO-ANCA) vasculitis type MPA with pulmonary and renal involvement, and a history of recurrent abortions and chronic pulmonary thromboembolism, being configured the diagnosis of APS due to the presence of antiphospholipid antibodies.
Clinical caseA 67-year old woman was admitted for study due to a clinical picture of 2 months of evolution of non-massive hemoptysis and subjective fever that appeared in the last month, with no other concomitant symptoms. At the beginning of the current clinical picture the patient was assessed in another institution, where they performed a chest CT-angiography, which revealed recanalized segmental thrombi and ground-glass areas both in upper and lower lobes, a diagnosis of pneumonia was established and it was decided to institute antibiotic treatment. A week later she was admitted to our institution. Her medical history was relevant due to the antecedent of hospitalization 2years before due to a picture of diffuse alveolar hemorrhage confirmed by fibrobronchoscopy and a transbronchial biopsy that revealed signs of pulmonary thromboembolism in histopathology, without evidence of capillaritis; given this, management with rivaroxaban was initiated (additionally she was receiving a supplement of levothyroxine for primary hypothyroidism). She was not taking other medications nor had other known diagnoses. Her obstetric history was important, since she reported 7 embryogenic abortions, with more than 3 that occurred consecutively before the week10. In the review of systems, numbness type paresthesias were found on the soles of both feet; other symptoms were negative.
At admission she was hemodynamically stable, with BP 104/65mmHg; HR 79/min; RR 20/min; SpO2 96% on ambient air; weight 74kg. The rest of the physical examination was only positive due to paleness in mucous membranes and fine crackles in the bases of both lungs.
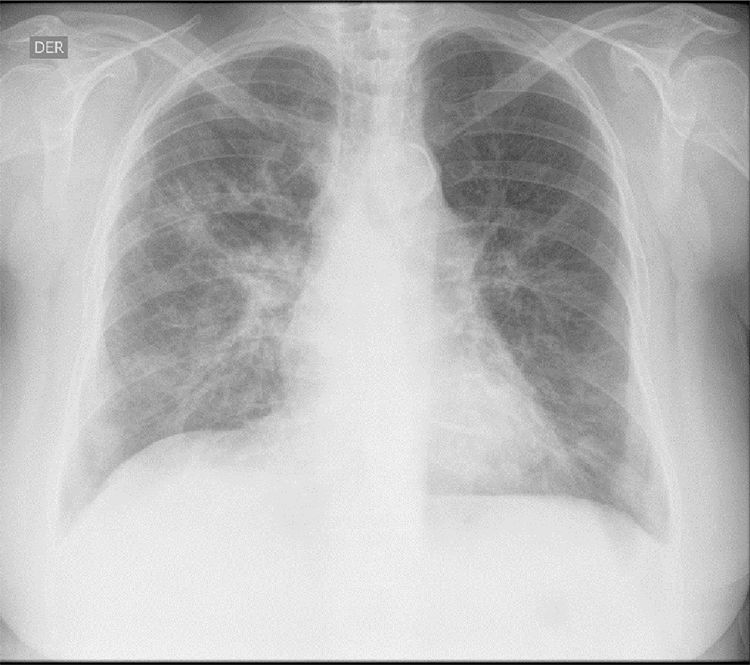
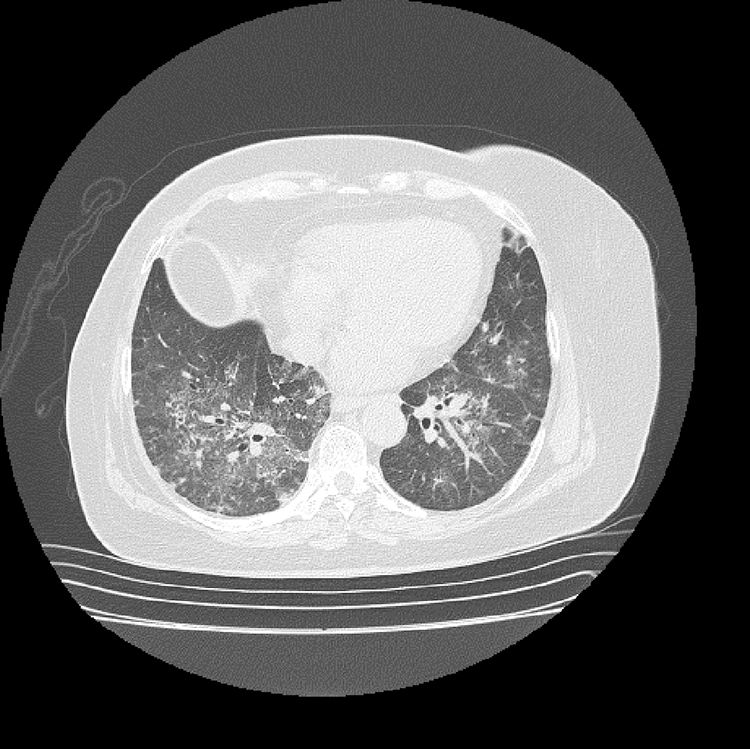
The initial laboratory findings revealed moderate anemia of normal volumes, elevated C-reactive protein (CRP) and active urinary sediment with proteinuria in the non-nephrotic range. The results of the diagnostic aids are shown in Table 1; the chest X-ray and tomography are shown in Figs. 1 and 2, respectively.
Results of paraclinical tests.
| Exam | Admission | Day 4 | Day 7 | Day 18 |
|---|---|---|---|---|
| Hemogram | Leukocytes 6460; neutrophils 4500; lymphocytes 1400; Hb 7.7; MCV 87.5; MCH 26.8; RDW 14.7; Hct 23%; platelets 443,000 | |||
| Coagulation | PTT: 32.7; PT: 14.7; INR 1.1 | |||
| Creatinine | 1.32 | |||
| BUN | 24.5 | |||
| CRP | 39 | |||
| NA | 142 | |||
| K | 3.48 | |||
| Urinalysis | Density 1020; proteins 150mg/dl; erythrocytes 250 (normal 0–5). Without erythrocyte dysmorphism. Bacteria+++ | |||
| Sputum smear | Negative for acid–alcohol resistant bacilli | |||
| 24h urine protein | 1760mg | |||
| Direct Coombs test | Negative | |||
| C3 | 163 | |||
| C4 | 24.8 | |||
| Rheumatoid factor | <8.0 | |||
| AG-HBS | Negative | |||
| ANTI-HVC | Negative | |||
| VDRL | Non-reactive | |||
| IgE level | 96.47 (high) | |||
| IgM level | 69 (normal) | |||
| IgG level | 1314 (normal) | |||
| IgA level | 319 (normal) | |||
| Anti-RO | <3.0 | |||
| Anti-LA | <3.0 | |||
| Anti-SM | 4.9 (negative) | |||
| Anti-RNP | 3.1 (negative) | |||
| Anti-DNA | Negative | |||
| ANCA | Positive 1/640 perinuclear pattern (ANCA-p) | |||
| Anti-myeloperoxidase (MPO) antibodies | 90.1 (positive) | |||
| Anti-proteinase 3 (PR3) antibodies | 0.64 (negative) | |||
| Anti-glomerular basement membrane (GBM) antibodies | Less than 1/20 | |||
| Antinuclear antibodies (ANA) | Negative | |||
| β2-Glycoprotein IgM | 1.4 (negative) | |||
| β2-Glycoprotein IgG | >100 (positive) | |||
| Anti-cardiolipin IgM | 6.6 (negative) | |||
| Anti-cardiolipin IgG | <3.0 (negative) | |||
| Lupus anticoagulant | Positive |


The patient was diagnosed with diffuse alveolar hemorrhage and glomerulonephritis. Anti-neutrophil cytoplasmic antibodies (ANCAs) by IIF were requested, which were positive in a titer of 1/640 with a perinuclear pattern, which was confirmed with the Elisa test, which was positive for myeloperoxidase (MPO) antibodies. A renal biopsy was performed with a finding of pauci-immune extracapillary proliferative glomerulonephritis and the electromyography demonstrated sensory polyneuropathy of axonal type in lower limbs, configuring the diagnosis of positive MPO-ANCA vasculitis type MPA (in the absence of granulomas) with high disease activity (BVAS 12/33 points). The antinuclear antibodies (ANAs) were negative and of the antiphospholipid antibodies, the anti-β2 glycoprotein 1 IgG resulted positive in a high titer, in addition to the lupus anticoagulant, the others being negative. Based on the obstetric antecedents, the finding of pulmonary thromboembolism by histology and chest CT angiography, in the presence of anti β2 glycoprotein 1 antibodies in a high titer, the diagnosis of APS was established, taking into consideration the need to repeat the antiphospholipid antibodies at 12 weeks, which were positive.
The patient was treated with intravenous pulses of methylprednisolone for 3 consecutive days, followed by oral prednisone at a dose of 1mg/kg/day. As maintenance therapy, rituximab was instituted, presenting a marked improvement in the renal and pulmonary function, with a decrease of the BVAS score to 2/33. Anticoagulation during the hospitalization was done with low molecular weight heparins, and warfarin was started prior to discharge with a target INR between 2 and 3.
DiscussionMPA is an ANCA-associated small vessel vasculitis. This group is completed by granulomatosis with polyangiitis (GPA) and eosinophilic granulomatosis with polyangiitis.4
The affectation in MPA is multisystemic; most patients have renal involvement; two thirds have musculoskeletal commitment; half have skin lesions; alveolar hemorrhage occurs in up to 30% of patients, and gastrointestinal and neurological compromise occurs in about one third of them.5
We describe the case of a patient with MPO-ANCA associated vasculitis that meets the characteristics of MPA according to the Chapel Hill consensus updated in 2012, with renal involvement given by pauci-immune extracapillary proliferative glomerulonephritis, demonstrated in histology; diffuse alveolar hemorrhage and peripheral neuropathy, in the absence of asthma, eosinophilia and granulomatous inflammation, which differentiates it from other ANCA-associated vasculitis.4,5
As in this patient, the ANCAs in MPA are predominantly directed against MPO, and their pathogenic role in this condition is recognized from numerous studies, although the mechanisms by which tolerance is developed and broken remain unknown.6 In a minority of cases the ANCAs may be directed against proteinase 3 (PR3), although their role in pathogenesis is less convincing.7
In the etiology of ANCA-associated vasculitis, both genetic and environmental factors (e.g., exposure to silica, drugs such propylthiouracil and infections, particularly by S. aureus) are recognized,8 none of these were identified in the patient.
In addition, chronic pulmonary thromboembolism was documented in this patient. The incidence of this disorder is high in patients with primary systemic vasculitis.9 This association has been studied more in patients with GPA; in the clinical trial The Wegener's Granulomatosis Clinical Occurrence of Thrombosis Study (WeCLOT), with a population of 180 patients with GPA, the incidence of venous thromboembolic events was 20-fold higher than in the general population.10 The reasons are not identified.
The presence of antiphospholipid antibodies is not an unusual characteristic in patients with vasculitis. A higher frequency of anti-cardiolipin and anti β2 glycoprotein1 antibodies at low titers has been found in patients with GPA; however, a causal association of antiphospholipid antibodies with thrombosis has not been demonstrated.11
In addition to GPA, antiphospholipid antibodies have been reported in association with polyarteritis nodosa, giant cell arteritis, polymyalgia rheumatica, Takayasu arteritis, Behçet syndrome, eosinophilic granulomatosis with polyangiitis and MPA.3
The study conducted by Rees et al.12 in 144 patients with primary systemic vasculitis, reported a prevalence of 17% of antiphospholipid antibodies; 6% (9 patients) met criteria for APS, and of the latter, 4 patients had ANCA-associated small vessel vasculitis (2 cases with GPA and 2 cases with eosinophilic granulomatosis with polyangiitis). No case of MPA was described, which is consistent with a search conducted in different databases.
The absence of capillaritis in the histological analysis of the lung specimen, taken during the first hospitalization of the patient, can be explained by a diffuse alveolar hemorrhage secondary to pulmonary thromboembolism. In this order of ideas and taking into account the obstetric antecedents of gestational losses occurred before the 10th week of pregnancy, associated with the laboratory criteria given by a positive result for lupus anticoagulant and anti β2 glycoprotein 1 IgG at high titers, the diagnosis of APS was confirmed.
In conclusion, we consider that the clinical spectrum of patients with MPA continues to be defined; the additional characteristics observed in our patient suggested an associated APS, which was finally confirmed. Therefore, monitoring the development of antiphospholipid antibodies in patients with primary systemic vasculitis and evidence of thrombosis or obstetric morbidity may be important in determining the need for prophylactic anticoagulation for prevention of thrombosis.
FundingNone.
Conflict of interestThere is no conflict of interest.
Please cite this article as: López-Villegas VJ, Medina-Morales DA, Saldarriaga-Rivera LM. Poliangitis microscópica y síndrome antifosfolípido. Una asociación infrecuente. Rev Colomb Reumatol. 2019;26:287–291.
