



Paget's disease of the bone is a metabolic bone disease of unknown origin, and is characterized by an increased phase of resorption, followed by an aberrant osteoformation phase. It is common in Europe, North America, New Zealand, and Australia, but infrequent in Asia, the Middle East, Africa, and in the Colombian population there are case reports. It is usually asymptomatic and is diagnosed incidentally by radiographic findings or an elevated alkaline phosphatase. The use of bisphosphonates favors the control of bone turnover and prevents complications such as fractures. A series of cases in Colombia is presented, along with a review of the literature.
La enfermedad de Paget ósea es una enfermedad metabólica del hueso de etiología no esclarecida, que se caracteriza por una fase de resorción aumentada seguida por una fase de osteoformación aberrante. Es frecuente en Europa, Norteamérica, Nueva Zelanda y Australia, pero infrecuente en Asia, Medio Oriente y África. En población colombiana hay reportes de casos. Generalmente cursa asintomática y se diagnostica incidentalmente por hallazgos radiográficos o fosfatasa alcalina elevada. El uso de bifosfonatos favorece el control del recambio óseo y permite prevenir complicaciones como las fracturas. Se presenta una serie de casos en Colombia y una revisión de la literatura.
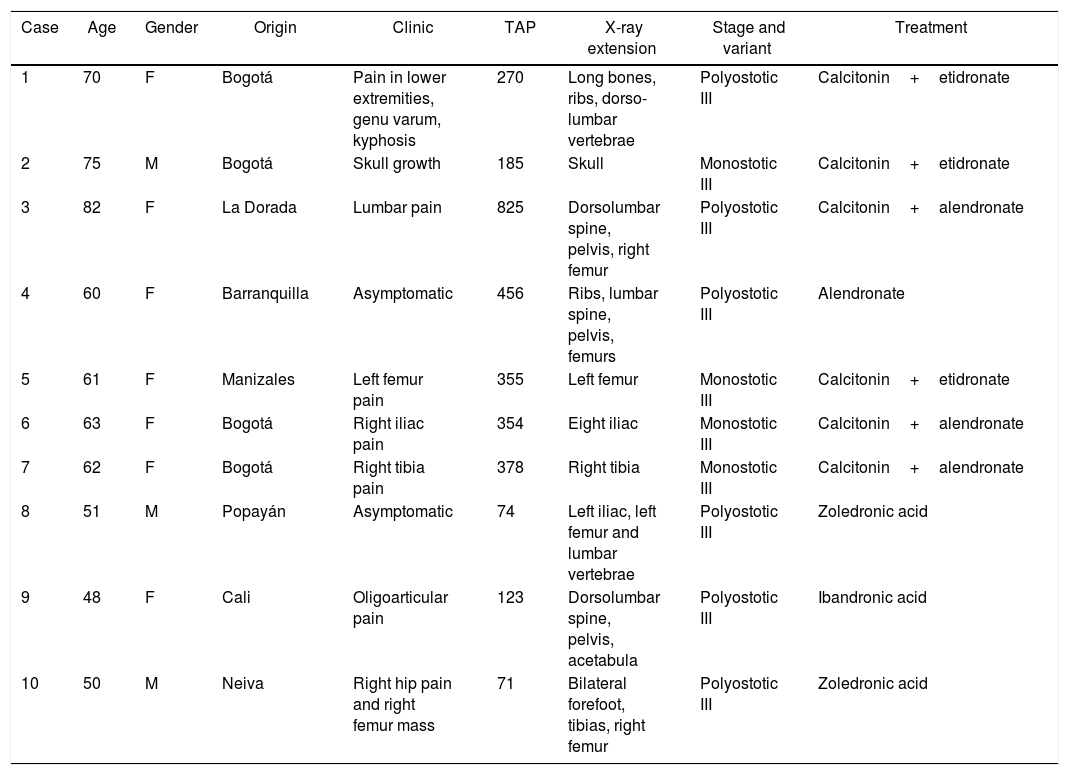
Paget's disease (PD) is a chronic osteometabolic disease, affecting one or several bones of the body. Its etiology is unknown, but influenced by genetic and environmental factors which play an important role in the pathophysiology of the disease. The highest prevalence is to be found in Europe, specifically in Great Britain, whilst it is very heterogeneous in South America, and is unusual in Colombia.1 Following is a discussion of a series of new cases in Colombia, which add up to the those previously published in our country2–4 (Table 1), and includes a literature review.
PD cases in Colombia and its main characteristics.
| Case | Age | Gender | Origin | Clinic | TAP | X-ray extension | Stage and variant | Treatment |
|---|---|---|---|---|---|---|---|---|
| 1 | 70 | F | Bogotá | Pain in lower extremities, genu varum, kyphosis | 270 | Long bones, ribs, dorso-lumbar vertebrae | Polyostotic III | Calcitonin+etidronate |
| 2 | 75 | M | Bogotá | Skull growth | 185 | Skull | Monostotic III | Calcitonin+etidronate |
| 3 | 82 | F | La Dorada | Lumbar pain | 825 | Dorsolumbar spine, pelvis, right femur | Polyostotic III | Calcitonin+alendronate |
| 4 | 60 | F | Barranquilla | Asymptomatic | 456 | Ribs, lumbar spine, pelvis, femurs | Polyostotic III | Alendronate |
| 5 | 61 | F | Manizales | Left femur pain | 355 | Left femur | Monostotic III | Calcitonin+etidronate |
| 6 | 63 | F | Bogotá | Right iliac pain | 354 | Eight iliac | Monostotic III | Calcitonin+alendronate |
| 7 | 62 | F | Bogotá | Right tibia pain | 378 | Right tibia | Monostotic III | Calcitonin+alendronate |
| 8 | 51 | M | Popayán | Asymptomatic | 74 | Left iliac, left femur and lumbar vertebrae | Polyostotic III | Zoledronic acid |
| 9 | 48 | F | Cali | Oligoarticular pain | 123 | Dorsolumbar spine, pelvis, acetabula | Polyostotic III | Ibandronic acid |
| 10 | 50 | M | Neiva | Right hip pain and right femur mass | 71 | Bilateral forefoot, tibias, right femur | Polyostotic III | Zoledronic acid |
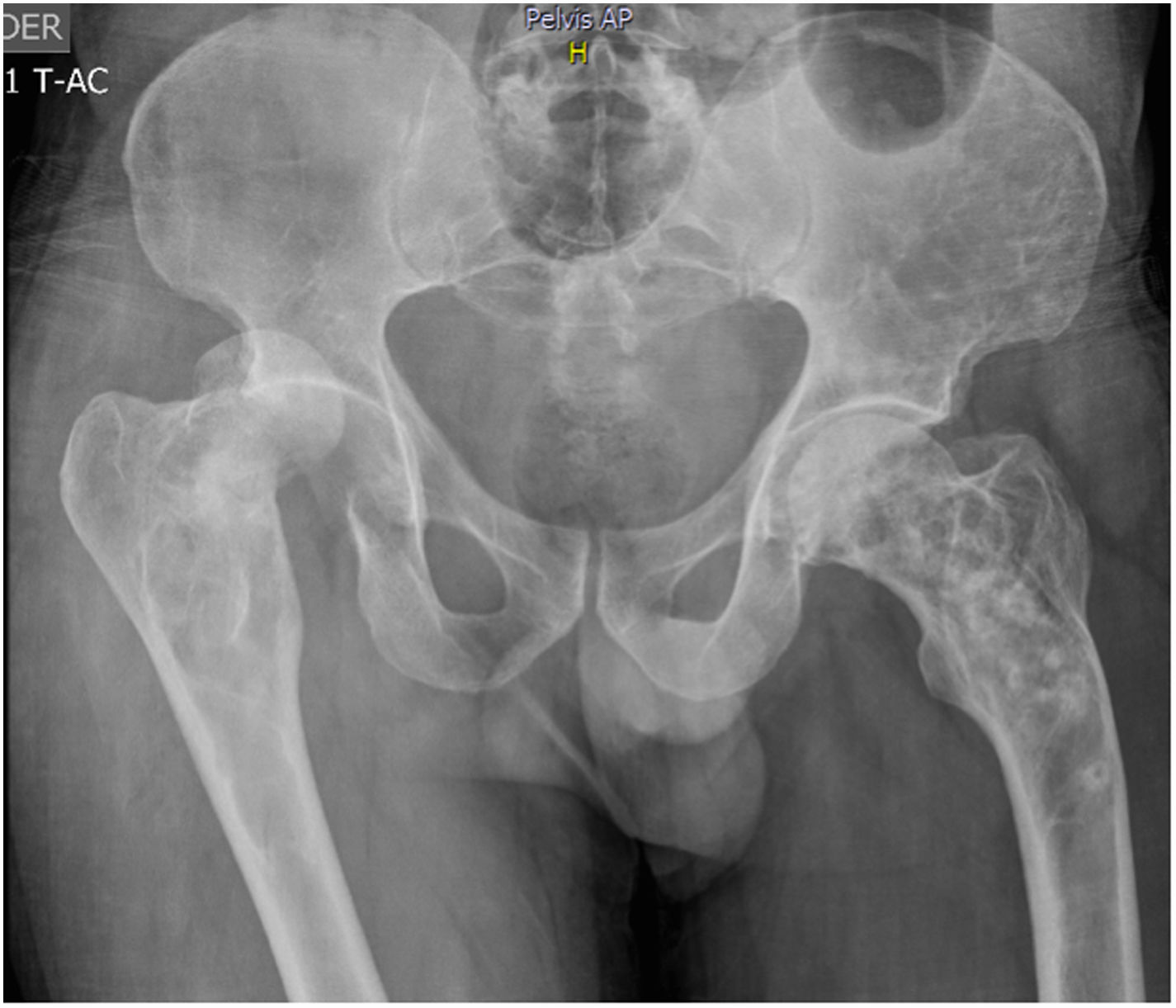
51-year old patient of Latin descent, with no personal or family history, who was previously asymptomatic, consults following a traumatic right hip dislocation. The plain X-rays incidentally identified: presence of osteolytic and osteoblastic alternating asymmetric lesions of the pelvis – left iliac wing – femur and spine, with L3 and L5 fractures. There were no clinical or paraclinical findings of prostate or GI tract cancer or myeloma. The CBC, renal and liver function were normal, the prostate antigen was normal, corrected calcium was 9.7; phosphorus 3.3; alkaline phosphatase 74, 25 (OH) vitamin D normal, thoracoabdominal CT-scan also normal. The patient was diagnosed with stage III polyostotic PD of bone, with spinal, pelvic and femur involvement. Zoledronic acid 5mg was prescribed with no complications. After one year of follow-up, the patient is asymptomatic, with no fractures or imaging changes (Fig. 1).
Case 2
50-year old male, Latin descent patient, with a history of gout, who visited the clinic with chronic right coccyx arthralgia associated with an ipsilateral femur mass. A bone lesion biopsy was performed revealing thickened and irregular bone trabeculae, mosaic pattern, basophilic cement lines, and normal trabecular alteration, suggestive of monostotic PD. The bone scan also showed abnormal osteogenic uptake in the bilateral forefoot, with a left predominance, increased uptake in the lateral tibial cortex suggestive of increased bone resorption. Additional paraclinical tests: alkaline phosphatase, serum calcium, serum phosphorus, 25 (OH) vitamin D, calciuria and phosphaturia in 24h, all within normal ranges. Single dose of zoledronic acid was indicated leading to hip pain resolution; for the underlying pathology, the treatment prescribed was febuxostat 80mg per day and colchicine 0.5mg per day.
Case 3Female, 48-year old patient of Latin ancestry, with a history of chronic kidney disease stage 5 due to nephrolithiasis and total left hip replacement due to arthrosis. She presents with oligoarthralgia of the large joints (shoulders, hips, knees) and lumbosacral mechanical pain.
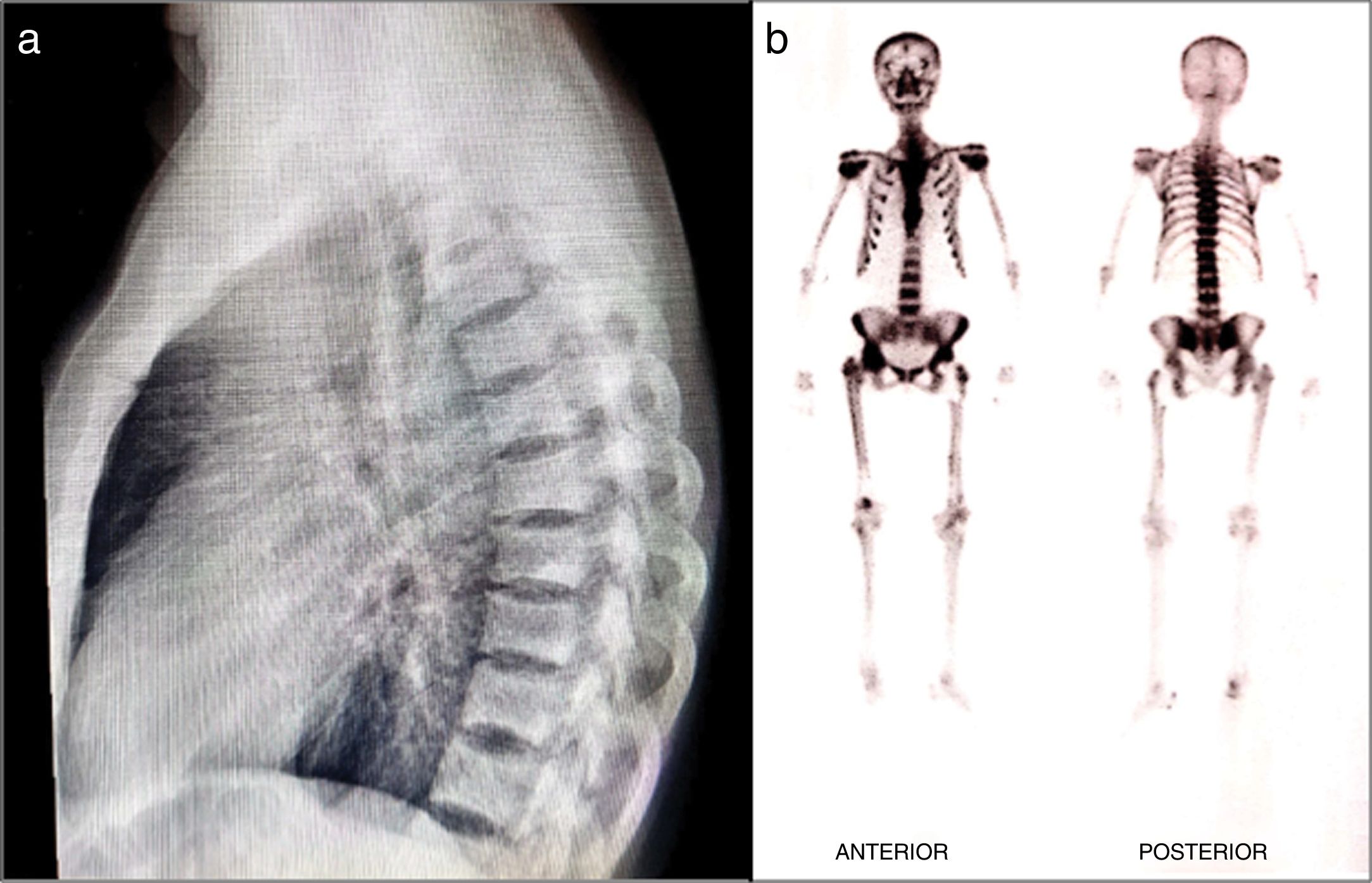
The physical examination revealed pain and limited mobility of the shoulders, hips and left knee, in addition to tenderness to palpation of the lumbar region. The X-rays showed areas with increased bone density in the vertebral bodies of the dorsolumbar and sacroiliac region and of both acetabula, in addition to changes due to arthrosis of the right coccyx. The bone scan showed increased uptake in the vertebral bodies, shoulders, hips, knees, and ankles. The T-score of the lumbar spine scan was 6.8 and in the femoral neck 1.2. The parathormone, calcium, phosphorus, and alkaline phosphatase serum levels were normal. The patient was then diagnosed with polyostotic PD and, due to the underlying renal involvement, the treatment prescribed was quarterly IV ibandronic acid for 5 doses. The tolerance was adequate, and complete osteoarticular pain resolution was achieved, and stabilization of the bone lesions according to plain radiograms and bone scans (Fig. 2).
Literature review X-rays: spinal osteoblastic lesions. (b) Bone scan: increased uptake lesions mostly in the axial skeleton, shoulders and pelvis of a patient with polyostotic PD III.")
PD is a metabolic bone disease of unclear etiology, characterized by a phase of increased resorption, followed by a phase of aberrant bone formation. The initial publications about the disease were made by Sancerote in 1801, Rullier in 1812, Wilks in 1869 and Czerney in 1873; but it was not until 1876 when the British surgeon pathologist James Paget described 5 cases, with the clinical and histopathological findings of the disease, which he called Osteittis deformans. He also described a form of intraductal breast cancer and a type of usually non-invasive adenocarcinoma of the skin, which are also named after him. In Colombia, the first case of PD was described in 1981 by Sánchez.5
EpidemiologyAround the world, PD is the second metabolic bone disease following osteoporosis, but with a very heterogeneous geographical distribution.6 There is a high prevalence among Northern European populations, with the highest reported in Great Britain, in the cities of Lancaster, Preston and Bolton (3–5% of the population over 55 years old), in areas of migration of these populations such as Australia, New Zealand, or the United States. The estimated prevalence in Spain is of 1.1–1.6%, with a higher prevalence in the extremely isolated rural areas exhibiting high rates of endogamy, as observed in several provinces neighboring the Central Plains (Sierra de la Cabrera in Madrid, 6.4%; Vitigudino in Salamanca, 5.7%).7,8 The disease is rare in the Scandinavian countries and Asia, with a prevalence of less than 1%. It is also rare in South America; however, large case series have been reported in Argentina and Brazil, probably because of a higher white European ancestry in these countries. In Colombia the disease is rare, with an approximate frequency of one case per 1,000,000 inhabitants.5
Between 5 and 40% of the patients have a family history of the disease, particularly in the high prevalence areas. The risk of developing PD in first degree relatives has been estimated at between 7 to 10-fold higher than in the general population. Some families have also been described in which the disease is inherited as an autosomal dominant trait, with high penetrance during the sixth and seventh decades of life.9 It affects both males and females, being slightly more frequent in males. It is a rare disease among people under 40 years of age. Its incidence increases with age, with estimates of 0.3 cases per 10,000 people-year in women and 0.5 cases per 10,000 people-year in males between 55 and 59 years old. This rate significantly increases after 85 years old, with 5.4 in females and 7.6 for males. The prevalence of the clinical diagnosis is of 0.3% for males and females over 55 years old.10
DP is a significantly old disease, even present in dinosaurs of the late Jurassic period (150 million years), diagnosed in one specimen in Tanzania.11
PathophysiologyPaget's bone lesions evidence increased osteoclastic-like bone resorption, accompanied by medullary fibrosis, increased bone vascularity, and increased bone formation, although irregular. Histologically, the lesions are labeled as mosaic, because of the mix of bone and lamellar tissue.12 There is osteoclastic increase with a larger number of nuclei and have nuclear inclusion bodies reminiscent of viral particles.13 However, the inclusion bodies are non-specific for PD, since they may also be present in other conditions such as osteopetrosis,14 picnodisostosis,15 and hereditary oxalosis.16
The rapid bone turnover in the disease causes bone production but with a disorganized architecture, poor mechanical strength, and high risk of deformity and pathological fractures. The disease is governed by a predominant osteoclastic action, by a high sensitivity to factors that promote bone resorption such as 1.25 dihydroxy vitamin D, and the NFkB (nuclear factor kB) ligand, also known as RANKL.17,18 Such association, particularly with RANKL, lead to consider that osteoclasts exhibit an apoptosis defect which extends their survival, but it has not yet been effectively demonstrated. Bone cultures of patients with PD compared against healthy controls, show a high sensitivity for osteoclastic promotion,19 as well as increased RANKL20 expression, with additional expression of IL-1, IL-6, Dickkop-1, that may further contribute to the focal bone turnover changes which are characteristic of the disease.21
The cause for PD it not completely clear yet, but some genetic factors have been identified as clearly predisposing for its development; at least 15% of the cases have been found to present a positive family background,22,23 and the risk of developing the disease increases between 7 and 10 fold among first degree relatives of patients, as compared against the general population.24 Studies in many families have allowed for the identification of an autosomal dominant disease, with a high prevalence in the sixth and seventh decade of life.25–27 Some mutations have been associated with regards to the development of the disease or related syndromes28; the most important for the development of classic Paget's is Sequestosoma 1 (SQSTM1), which codes for p62 (“scaffold” protein), linked to NFkB signaling.29 The alteration of the domain associated to the ubiquitin in SQSTM1 has been reported as the cause for 20–50% of all familial cases, and of 5.2% of the isolated, sporadic cases.24,30–32 Such mutation, in contrast to those in which the mutation is absent, allows for the development of a more extensive disease, and with a prevalence between 79 and 100% in the seventh decade of life. Another genetic candidate linked to the development of PD, is in the 5q31 and 10p13 chromosomes, although the disease-associated genes have not yet been identified.25,26
The mechanism responsible for the development of the disease is not clear either. It has been considered that factors such as mechanical stress is a potential cause for bone lesions, and hence its predominant presentation in the axial skeleton and lower extremities33 Some environmental factors have been described as potential triggers of PD. Observational studies and hypothesis include: low calcium and vitamin D intake during childhood,34,35 occupational exposure to toxins,36 zoonotic37 and viral38 infections, of which the most broadly studied is paramyxovirus infection, particularly the canine virus distemper and measles virus,32,39,40 given the aspect of nuclear inclusion bodies recalling the presence of viral nucleocapsids,13 although the results have been conflicting and poorly conclusive.41–44 The in vitro effect has been demonstrated on the increased number of osteoclasts and bone turnover, but is has not yet been correlated in vivo.45
Clinical manifestations and complicationsThe clinical manifestations of the disease are quite variable and range from an asymptomatic presentation up to situations with bone pain and complications such as deafness, deformity fractures, and predisposition to develop neoplasms. The asymptomatic presentation is the most frequent, and hence most diagnoses are incidental. 5
Bone painPain is the most frequent symptom and prevails in the lower extremities (lumbosacral region, hips and legs). The cause of pain is not clearly elucidated yet, but may be due to distension of the periosteum at the bone formation sites that compress nerve endings. Some patients evolve to the osteolytic phase and also complain about pain, whilst others despite the bone formation do not express any pain; hence this symptom is a frequent occurrence, but of variable or unknown etiology. In general, pain improves with mobility and worsens with rest, unless in the presence of secondary osteoarthosis, juxta-articular to Paget's lesions. The pain may also be secondary to complications such as pathological fractures, fissures or deformity, with secondary osteomuscular and mechanical imbalances.5
The most frequently affected bones are the pelvis, the femur, the spine, the skull and the tibia, but the sternum, the clavicles and ribs may also be compromised. The lesions that develop at the onset continue to be present over time and it is unusual for new lesions to develop during follow-up; however, when the affected bone is transplanted, new lesions may develop in the recipient bone. The lesions in the long bones initiate in the proximal region of the epiphysis and progress along its axis at a rate of 8mm/year. The maximum point of encroachment of the lesion looks like a V-shape wedge, reflecting the osteoclastic resorption and increases the risk of fracture. The same phenomenon is seen in the skull, as a circumscribed osteoporosis.46 After the bone resorption phase, the bone formation phase follows, which evidences an increase in the anteroposterior diameter of the skull, more pronounced at the frontal and occipital level. Facial bone involvement is unusual. There may be some dental involvement with obliteration of the lamina dura and formation of bone cement at the root of the nerve, and there may be some anchyloses.5
Neurological complicationsWhen the otic capsule is compromised in the skull, neurosensory hearing loss may develop, and patients may present with tinnitus or vertigo.5,46 Myelopathy per se is rare and may develop due to vascular steal rather than as a result of compression.
FracturesFractures may occur during the lytic phase due to demineralization or during the mixed phase due to non-laminar bone matrix deposits; the most frequent sites are the tibia and the femur.
DeformityDue to fragility and loss of the normal bone architecture, there is bowing of the long bones, upper third of the tibia, femur, and consequently coxa varum, protrusion of the acetabulum, coxa and secondary gonarthrosis.
Malignant transformation of Paget's DiseaseThis is a rare but severe complication. In most cases, the tumor is an osteosarcoma, but there is a subgroup in which giant cell tumors develop. The latter is a familial syndrome and is associated with ZNF687 gene mutations. These gene codes for a C2H2 zinc finger protein, which is part of a transcriptional regulatory complex that expresses in most tissues, including the bone. The disease in these patients is usually polyostotic and of early onset. The tumors are multifocal and appear after 10 years of the disease, but the interval may vary from 3 to 30 years. This alteration is more prevalent in Southern Italy, a population in which the mutation has been detected.46
DiagnosisThe fundamental diagnosis is radiological and often incidental, as a result of the asymptomatic evolution of most patients. Plain X-rays show the thickening of the cortexes, circumscribed osteoporosis, alternating sclerotic or lytic images, loss of the cortico-medullary differentiation, and increased bone size. It also depicts the presence of complications such as deformities and fractures.47 In the study of new areas of pain on which a plain X-ray is not conclusive, it is possible to use CT, PET/TC with radiotracers such as for instance F18-sodium fluoride, or MRI.48 The bone scan has a limited specificity for diagnosis, and although it may be used to assess the extent of the disease – for which it is very sensitive – identifying any pelvic or vertebral involvement using X-rays is already a determinant for antiresorptive therapy, and hence in this case such test may be avoided.
Based on the radiographic findings of the disease, it may be classified as “active” or “inactive”. The former is further subdivided into early – stage I or osteolytic, which affects the skull and long bones with subchondral circumscribed osteoporosis with encroaching wedge-shape radiolucency. Early – Stage II or osteolytic/osteosclerotic/both, affecting the skull, the long bones and the pelvis, with the changes described for Stage I, in addition to patched radiolucency and radiodensity compromising the diaphysis, the epiphysis, and metaphysis. The late, inactive, stage III or osteosclerotic category includes skull, long bones, pelvis and spine involvement; in these cases, the cotton wool appearance prevails, with thickening of the cranial dome and pelvic bones, with focal or diffuse radiodensity, picture frame or ivory vertebral bodies, epiphyseal predilection, trabecular thickening, bone widening and deformity.5
From the serological point of view, the incidental finding of elevated total alkaline phosphatase (TAP), in relationship to elevated levels of gamma glutamyl transpeptidase (GGT), indicate hepatobiliary pathology. In contrast, normal GGT levels in an adult, non-pregnant individual, are probably due to bone etiology. TAP es elevated in 85–95% of the patients with untreated PD, it is correlated with the disease activity and therefore may be used as a follow-up biomarker. Normal alkaline phosphatase levels are present in monostotic disease, few bone affected, or metabolically inactive disease.49
Measuring other bone formation markers such as osteocalcin and procollagen type I peptides and bone resorption markers, including pyridinolines, hydroxyproline and reticular collagens, fail to provide any additional information, are non-specific and the diagnosis is better established using images.1,50 Nonetheless, some authors claim that N-propeptide procollagen-1 seems to work better by rising above the normal level, even if it is a limited extension disease.51 Measurement of urinary hydroxyproline was used for many years; however, it has been abandoned because it is a burdensome test with limited sensitivity and specificity.
Very rarely, a bone biopsy is required to make the diagnosis, although it may be helpful to differentiate from osteoblastic metastasis or osteosarcomas if the clinic is suggestive. Histologically, the bone microstructure is highly abnormal in PD. Osteoclasts increase in number, size, and nuclearity, and present nuclear inclusions. Initially it was thought that these inclusions were paramyxovirus, but more recently it has been suggested that they are aggregates of non-degraded proteins due to defects in the autophagic pathway. Bone formation also increases in response to increased bone resorption. However, the newly formed bone is abnormal and is deposited in a chaotic manner, resulting in a mechanically weak bone. Other histological characteristics of active PD include increased vascularization and medullary fibrosis.52
Histological analyses such as histomorphometry of the vertebrae and the pelvis has been used in research studies and confirm greater trabecular thickness, high bone turnover with a significant increase in bone resorption and bone formation indexes (number of trabeculae, osteoid volume and osteoid surface, number of osteoblasts and osteoclast surface), and increased bone volume. However, it is claimed that there is a better correlation between bone formation and bone resorption in osteoporosis.10
Differential diagnoses: Congenital, degenerative, infectious (chronic osteomyelitis), metabolic (rickets, osteomalacia, hyperparathyroidism, renal osteodystrophy) diseases; traumatic or malignant (primary due to osteosarcomas or metastatic particularly due to prostate cancer and myeloma; diseases such as fibrous dysplasia).46 In these cases, the information based on the medical record of associated symptoms and altered laboratory test findings is valuable.
TreatmentThe treatment for PD currently gives rise to some controversy. 3 main reasons have been suggested to implement pharmacological treatment. The first, from the pathophysiological point of view, is aimed at normalizing bone turnover as much as possible.53 The second reason is clinical for pain control; and the third approach is intended to treat complications. These three basic objectives may generally be achieved with the use of bisphosphonates, which have proven their effectiveness in reducing bone turnover and controlling pain, but are apparently less effective in preventing several complications of the disease.
The use of pain killers, initially with paracetamol and then other more potent analgesic agents, in many cases provide adequate pain control from PD. However, the long-term safety of analgesics and the lack of effect on the pathophysiology of the disease, lead us to look for other therapeutic options. Historically, calcitonin was one of the first agents used to control PD because of its activity on bone turnover.54 However, its short-lasting action very promptly led to its replacement by bisphosphonates and its use nowadays is increasingly rare.55
Different bisphosphonates have been used in the treatment of this disease. Nowadays, more potent and longer-acting bisphosphonates are available. They have shown in various trials (some randomized, controlled trials), not just bone pain control, but also additional effects, such as reducing bone turnover, and restoring bone quality.56 The discussion has focused on length of use and its ability to prevent other complications such as hearing loss or nerve compression. In terms of treatment duration, different regimens have been adopted, depending on the type of bisphosphonate used. However, the risk of relapses following its discontinuation prompted research for more adequate monitoring and treatment protocols. Pamidronate, after an initial IV dose of 90mg (divided into sessions of 30mg per day), may have an effect lasting up to approximately 12 months. Other bisphosphonates such as alendronate (not endorsed in several European countries) or risedronate, may have an effect lasting up to 5 years, but using daily doses for several months. Other drugs of this type, such as etidronate, tiludronate, etc., are less frequently used. At present, zoledronate is considered to be the most potent and longer-lasting bisphosphonate, with the potential of achieving clinical response and bone turnover impact for more than 6 years, after one single IV dose.1
When using the TAP serum levels to monitor the effect of these drugs, Zoledronate has shown a 75% reduction of this enzyme in 98% of the patients for 6 months. Relapses (defined as a TAP elevation of more than 20% of the pre-treatment levels) only developed in less than 1% of the patients after 6.5 years of observation.57,58 With regards to the capacity of bisphosphonates to avoid other complications described, the PRISM trial failed to show a significant difference between the use of bisphosphonates and the use of analgesic agents, showing that the rate of long-term complications was similar, without evidencing a higher reduction with the use of bisphosphonates. Nonetheless, it is also possible that bisphosphonates are effective in a rare complication due to paraparesis as a consequence of vertebral involvement.59
The use of denosumab has been recently suggested, which could theoretically be an attractive treatment option, but its use so far has only been incidental, though with apparently encouraging results.54,60
Finally, for the treatment of mechanical complications associated with PD-related bone deformities, orthopedic surgery has been considered. However, due to the greater bone vascularization present in this disease, there is an increased risk of bleeding over the course of surgery. For this reason, pre-treatment regimens with bisphosphonates have been used, in order to reduce the risk of bleeding or loss of the prosthesis due to accelerated bone turnover. In any case, previous medical treatment necessarily delays surgery by a few months.61 Other complications such as hearing loss, medullary compression as a result of vertebral lesions, or cardiac failure have also received bisphosphonate treatment with poor response.
DiscussionPD is an osteo-metabolic disease, highly prevalent in Europe and in areas with a high number of European immigrants. However, it is very rare in Asia, the Middle East and Africa. In Latin America, the distribution is heterogeneous, and particularly in Colombia, only sporadic cases continue to be reported. Adding the 3 cases herein reported to those described by Sánchez et al.,2–4 there is a total of 10 cases, all of them over 40 years old, which is the age from with the patients in the world literature have been reported. Notwithstanding the fact that the disease affects both genders similarly, 70% of the cases we have reported are females but we have been unable to establish whether there is really a hormonal or chromosomal factor involved, that favors such difference, as it is the case with osteoporosis. Likewise, the mechanisms responsible for the development of the disease, such as mechanical stress, infectious and nutritional factors, do not show overt differences between the two genders in our setting. None of the 3 cases described have any family history of PD, which suggests that beyond the genetic factors, in our setting we should consider multiple environmental factors which are not clearly known, as potential drivers for the disease.
The clinical manifestations were the ones described worldwide, without any neurological complications, fractures, deformities or malignant transformations. Notwithstanding the fact that some patients underwent extension studies, in every case the diagnosis was made based on typical plain X-ray findings. Six of them with polyostotic disease, all inactive, late or stage III, with particularly sclerotic changes as expected. Three patients had normal TAP, which is particularly likely in cases of inactivity as is this one.46
The reasons for treating the patients were to normalize bone turnover, modulate pain and avoid complications from neurological compression in patients with vertebral involvement, considering that the literature claims that treatment may minimize the risk of paraparesis.59 Historically, calcitonin, was one of the first medications to be used to control Paget's Disease, but due to its short-lasting action, it was replaced by bisphosphonates, in particular zoledronic acid as a single dose, since it helps to minimize the relapses of the disease for longer periods of time.56 In the cases just reported, bisphosphonates were used.
ConclusionsPD is a chronic, osteo-metabolic disease with a heterogeneous geographical distribution. In our setting, its presentation is asymptomatic or with bone pain, both in males as in females over 40 years old. The diagnosis is essentially radiographic, and treatment is based on the use of bisphosphonates, particularly when intended to control pain; it is not yet clear if it has an impact on reducing the risk of complications, particularly of neurological compression when vertebral involvement is present.
Conflict of interestsThe authors have no conflict of interests to disclose.
Please cite this article as: Ospina Caicedo AI, Gómez Escobar VE, Coy Urrea VA, Segura Charry JS, Izquierdo Loaiza JH. Enfermedad de Paget de hueso esporádica. Serie de casos y revisión de la literatura. Rev Colomb Reumatol. 2020;27:103–111.
