




Stiff person syndrome affects the central nervous system. Relevant clinical signs are stiffness, muscle spasms, increased sensitivity with external stimuli that increase muscle contractions. Women are affected twice to three times more, in comparation with the men. There are characteristic clinical and electrophysiological type markers. The etiology is associated with mediation by antibodies and may be the expression of a paraneoplastic syndrome. Pharmacological treatment is focused on muscle relaxant-type medications, drugs with immunomodulatory or immunosuppressive mechanism. In addition, complementary rehabilitation treatment is required.
The purpose of the group is to make the description of the clinical case that is relevant due to the low frequency of presentation and to carry out an update of the topic.
El síndrome de persona rígida afecta el sistema nervioso central. Los signos clínicos relevantes son la rigidez, los espasmos musculares y sensibilidad incrementada a los estímulos externos, que inducen las contracciones musculares. Las mujeres son afectadas de 2 a 3 veces más con relación a los hombres. Hay marcadores de tipo clínico y electrofisiológico característicos. La etiología se asocia con la mediación por anticuerpos y puede ser la expresión de un síndrome paraneoplásico. El tratamiento farmacológico se realiza con medicamentos relajantes musculares y medicamentos con mecanismo inmunomodulador o inmunosupresor. Adicionalmente, se requiere un plan complementario de rehabilitación.
El propósito del grupo es hacer una descripción del caso clínico, que consideramos es relevante por su baja frecuencia de presentación y realizar una actualización sobre el tema.
The stiff person syndrome (SPS) is a disease affecting the central nervous system and manifests with stiffness, muscle spasms, and increased sensitivity to external stimuli that further deteriorate the contractions. There are distinctive clinical and electrophysiological markers such as the simultaneous contraction of agonistic and antagonistic muscles, the involuntary and continuous motor unit firing at rest.1–4
The reports submitted indicate a prevalence of 1–2 cases per million per year. The mean age at presentation varies between 20 and 50 years, women are more frequently affected in a proportion of 2–3 times more than men.3,4
The goal is to make a clinical case description that we feel is relevant on account of the low frequency of presentation and do an update on the topic.
Clinical caseThis is a female patient who presented with varying symptomatology back in 2012, including “electric shock” type pain in the extremities, in addition to involuntary facial movements, difficulty to articulate words, distal weakness of the lower limbs associated to stiffness, focal painful muscle contractions of several muscle groups that worsened with environmental stimuli; subsequently, the effect became generalized up to the development of lack of motor coordination, slow gait, and marked feeling of fatigue. The physical examination revealed severe dysarthria, hypotonic tongue with limited protrusion, head tremor, bilateral dysmetria and ataxic gait. In accordance with the presentation of the clinical characteristics, a probable diagnosis of SPS was considered, and hence the relevant tests were prescribed in order to elucidate the potential etiology (Table 1). The metabolic profile tests, as well as infection screening, nutritional, occult neoplasm and immunology analyses were all reported as normal. The presence of anti-glutamic acid decarboxylase antibody (GAD) was specifically screened for, and the report was negative.
Extension studies.
| Cerebral | Small hypersensitivity areas with non-specific white matter punctiform signals. |
| Brain spectroscopy | Spectral pattern alteration identified, with increased concentrations of glycine or myoinositol found on the same spectral position (3.56) at the level of the right basal ganglion, with no further abnormalities in the spectral patterns or in the relationships of the metabolites assessed. |
| Spine | Mild osteochondritis-type changes in the cervical spine, central osteophyte at C3–C4 which partially obliterates the anterior subarachnoid space; additional findings were identified at the level of T7–T8 and T8–T9. |
| Functional language | Left language dominance with very mild activation with different types of paradigms. Presence of motor activation of the tongue usually localized in the bilateral precentral gyrus. No alterations of the tracts studied and the t1 volume study did not show any evidence of atrophic changes. |
| Electroencephalogram | Normal |
| Echocardiogram | Normal |
| Repetitive muscular stimulation test | With no electrophysiological evidence of presynaptic or postsynaptic disorder of the neuromuscular junction. |
| 4 extremities nerve conduction studies | Motor and sensitive nerve conduction in the 4 extremities (medial, ulnar, tibial, fibular and sural) resulting in latencies, amplitudes and conduction velocities within the normal limits. |
The comprehensive management of the patient included symptomatic pharmacological therapy with buspirone, levodopa–carbidopa, pregabalin and a rehabilitation plan with speech therapy for improving swallowing and voice, physical therapy with emphasis on hydrotherapy for improved mobility and independence.
The 2-year follow-up showed a progressive loss of independence to carry out the activities of daily living and increased compromise of gait, which required the prescription of a wheel chair. By the year 2017 the clinical condition further deteriorated and the patient had to be permanently assisted to conduct the basic activities of daily living until she was finally hospitalized. On this occasion, the physical examination revealed an alert patient, with severe dysarthria and naming difficulty; the patient could not hold her head up or keep her balance in the sitting position. The muscle examination revealed exacerbated myotendinous reflexes, hypertrophic hypertonic muscle groups, with involuntary contractions, sustained muscle spasms and extreme difficulty for relaxation, causing significant pain.
The case was studied by the group of treating physicians, with the following findings:
- 1.
A constant and slowly progressive evolution throughout the years of the disease, progressing toward functional disability.
- 2.
The clinic showed stiffness, generalized and painful muscle spasms exacerbated by environmental impacts.
- 3.
The electrodiagnostic studies recorded continuous involuntary activity, with no evidence of peripheral nervous system involvement.
- 4.
Absence of cachexia or any other signs of malnutrition.
- 5.
The haemato-oncology team conducted paraclinical extension studies for occult neoplasms, including bone marrow biopsy, flow cytometry for immature forms of lymphocytes which was negative; the immunoglobulin serum levels measured were normal, making it unlikely to diagnose cancer or paraneoplastic syndrome. Consequently, the antineuronal autoantibodies profile was not considered a requirement.
The conclusion was that the clinical condition of the patient was probably consistent with SPS of probable autoimmune etiology.
The studies were updated (Table 2) and the etiological therapy suggested was plasmapheresis and complementary symptomatic treatment with baclofen. The patient remained hospitalized until completion of 10 plasmaphereses, evidencing a clinical improvement after the seventh exchange procedure. This improvement materialized as a reduction in dysarthria, less muscle spasms and improved muscle tone, which improved the patient's mobility and trunk control. Additionally, low vitamin-D levels were documented, but despite supplementation, there was no evidence of improvement in muscle stiffness upon normalization of the vitamin-D levels. The patient was discharged with pharmacological therapy based on baclofen, buspirone, levodopa–carbidopa, pregabalin, vitamin-D and a rehabilitation program with speech therapy for improving swallowing, and dysarthria, and to improve the patient's ability to communicate. Additionally, she was prescribed physical and occupational therapy to maintain the articular rage of movement, modulate muscle tone, improve balance, coordination and motor control.
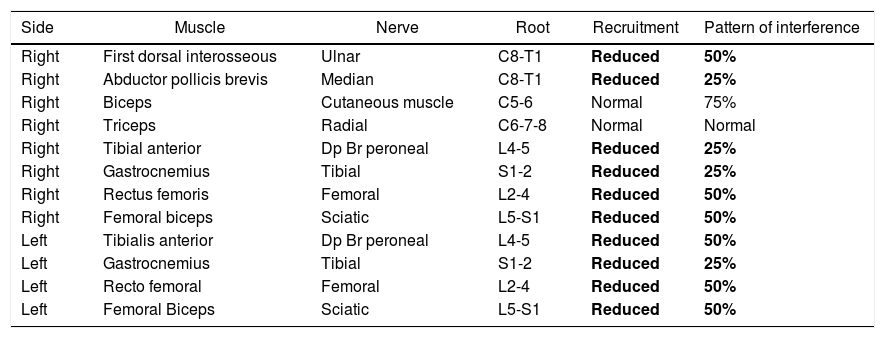
Electromyography.
| Side | Muscle | Nerve | Root | Recruitment | Pattern of interference |
|---|---|---|---|---|---|
| Right | First dorsal interosseous | Ulnar | C8-T1 | Reduced | 50% |
| Right | Abductor pollicis brevis | Median | C8-T1 | Reduced | 25% |
| Right | Biceps | Cutaneous muscle | C5-6 | Normal | 75% |
| Right | Triceps | Radial | C6-7-8 | Normal | Normal |
| Right | Tibial anterior | Dp Br peroneal | L4-5 | Reduced | 25% |
| Right | Gastrocnemius | Tibial | S1-2 | Reduced | 25% |
| Right | Rectus femoris | Femoral | L2-4 | Reduced | 50% |
| Right | Femoral biceps | Sciatic | L5-S1 | Reduced | 50% |
| Left | Tibialis anterior | Dp Br peroneal | L4-5 | Reduced | 50% |
| Left | Gastrocnemius | Tibial | S1-2 | Reduced | 25% |
| Left | Recto femoral | Femoral | L2-4 | Reduced | 50% |
| Left | Femoral Biceps | Sciatic | L5-S1 | Reduced | 50% |
The needle electromyography showed sustained, irregular and paroxysmal involuntary muscle activity of multiple muscle groups with impossibility to accomplish rest or absolute relaxation, with absent signs of membrane instability and of neuromyotonic discharges.
Bold letter: highlights the abnormal findings of the electromyography study.
Finally, the decision was made to discontinue plasmapheresis and continue outpatient treatment with rituximab. This decision was supported by the positive reports discussed in the medical literature, the easy dosing and administration of the product. The patient was administered an IV infusion with 1000mg and an additional dose after 15 days. The regimen was repeated over the next 4 months, and in total she has received 3 cycles. Clinical follow-up was conducted. In contrast to the clinical condition reported in 2017, the patient improved in terms of the intensity of the axial and cervical muscle spasms that caused significant dysphagia, the sitting balance and independence in daily life activities also improved, and she was able to move around with a walker and therefore she now enjoys a better quality of life.
DiscussionThe etiology of SPS is associated with antibody mediation that, when combined with antigens, result in functional block of the neuronal synapses – in the brain and spinal cord – that use gamma aminobutyric acid (GABA)1 as neurotransmitter. The best reported antibodies are: anti-GAD antibodies – glutamic acid decarboxylase (GAD) autoantibody – antiglycine alfa 1 (anti-GlyR) antibodies, antianphiphysin antibodies and gephyrin.1 The synthesis to produce GABA has 2 isoforms: the antibody formation against the GAD65 isoform which is the most frequently reported in patients with SPS, localized in the presynaptic terminal and anchors in a reversible manner onto the membranous synaptic vesicles. The pathogenic effect of the anti-GAD65 antibodies is uncertain, due to the intracellular localization of the antigen which hinders antibody action and expresses with different clinical syndromes.1–3 The anti-GAD antibodies are not specific for SPS, since up to 1% of the normal population assessed expresses them, are positive in up to 5% in different neurological syndromes such as: cerebellar ataxia, limbic encephalitis with myoclonus and temporal lobe epilepsy.1,2 The clinical severity of the disease is unrelated to the level of antibody titers measured in serum and in the CSF; there are reports of patients who develop a severe disease with low titers and vice versa, whilst others who have the disease have negative serum titers.1,5
Antianphiphysine and anti-gephyrin antibodies – synaptic proteins – are expressed in paraneoplastic syndromes associated with breast and ovarian tumors, small-cell lung cancer, renal cell carcinoma, thyroid carcinoma, colon cancer, thymoma, Hodgkin's and non-Hodgkin lymphoma, colangiocarcinoma.1,3 In SPS, as in the paraneoplastic syndromes, a defective endocytosis of the transmitter develops caused by the antibody.1 In the case of SPS in cancer patients, a direct pathological mechanism of the antibodies is suggested, which was shown in laboratory experiments with mice that were infused with specific antianphyphysine antibodies, which developed a clinical syndrome, with abnormal electromyography studies and positive central nervous system antibodies.1 T and B-cells involvement is not clear. The assumption is that the microglial cells and B-cells function as T-lymphocyte antigen presenting cells (GAD65 antigen), hence perpetuating their impact on the central nervous system, and B-cells are linked to the generation of oligoclonal populations of antibodies.3,5
Genetically speaking, different genes of the major histocompatibility complex (MHC) are expressed in SPS – particularly the DQB1*0201 and DRB1 alleles of CMH II.1,3
The clinical presentation of the disease was classified in subtypes, as follows3,4,6:
- •
Classical type SPS
- •
SPS variants
- •
Focal o segmented
- •
SPS with spasms
- •
Variant of progressive encephalomyelitis with stiffness and myoclonus.
- •
SPS with ataxia, epilepsy, etc.
- •
Paraneoplastic variant
The progressive encephalomyelitis variant with stiffness and myoclonus, affects people between 50 and 60 years old, with an insidious onset and evolution with a pattern of remission and relapse; there is brain stem dysfunction, signs of dysautonomia, and is related to anti-glycine alfa 1r (anti-GlyR) antibodies.3
For the SPS diagnosis – classical variant – the criteria submitted are3,6–8:
- •
Inexpressive facies with stiffness of the axial muscles and extremities, predominantly in the abdominal and thoracolumbar paravertebral muscles which cause a constant hyperlordosis-like deformity, with difficult breathing due to the involvement of the thoracic muscles.
- •
Painful muscle spasms caused by environmental stimuli such as noise, emotional and tactile stresses. The spasms are intermittent and similar to those present in tetany, last several minutes and may result in bone fractures or joint dislocations.
- •
The electromyogram shows continuous motor activity in the agonistic and antagonistic muscles.
- •
Compromise due to other neurological diseases should be ruled out.
- •
Positive anti-GAD65 or antianphiphysine serum antibodies.
- •
Clinical improvement with benzodiazepines.
Fatigue in patients can be accounted for by the presynaptic defect at the neuromuscular junction as shown by electrophysiological studies.9
SPA should be differentiated from pathologies compromising both the pyramidal and the extra-pyramidal system, for instance: anti-N-methyl-d-aspartate (NMDA) receptor encephalitis, limbic encephalitis, refractory epilepsy, myelopathies, dystonia, spinocerebellar degeneration, primary lateral sclerosis, neuromyotonia, tetanus, myasthenia gravis, type I diabetes (in 35% of the cases coexists with the syndrome), autoimmune thyroiditis, Graves’ disease, polyendocrine autoimmune syndrome, vitiligo, retinopathy and autoimmune scleritis, lupus, pernicious anemia and celiac disease.1–3
The diagnostic methods include brain and spinal cord MRI, electromyography with the above described findings and electroencephalograms.1–4 A complete test screening should be conducted to cover the above-mentioned broad differential spectrum. In case of patients suspicious of the paraneoplastic variant, the Anti-Ma, Anti-Yo, Anti-Hu, antianphiphysine and gephyrin paraneoplastic antibodies shall be measured, as well as the anti-acetylcholine receptor, anti-glycine receptor, anti-GABA receptor, anti-GAD receptor.10
The treatment is addressed in accordance with the positivity of the antibody identified as positive; there is evidence of patients with positive antibodies against the GABA receptor which respond better to immunoglobulin. The anphiphysine positive patients respond better to steroids, plasmapheresis, and primary cancer treatment; the anti-GAD positive responded well to immunoglobulins, diazepam, clonazepam, whilst the anti-GlyR alfa 1 positives have improved results with immunotherapies.8
With regards to the SPS pathogenesis, the following therapies are available3,4,7:
- •
GABAergic medications: benzodiazepines, botulin toxin, intrathecal baclofen with average results. In order to modulate symptoms, other options such as gabapentin, tiagabine, valproate, levetiracetam, are suggested.7,8
- •
Medications with immunomodulatory o immunosuppressive effects. The best reports include:
- ∘
Prednisone.
- ∘
Plasmapheresis: Dra. Pagano et al. reported their experience with a group of 9 patients treated with plasmapheresis who had worsened with first line therapies; 78% reported a mild clinical improvement and 56% experienced a significant improvement, with good tolerance to treatment. The study by Johns Hopkins Institute found positive results with plasmapheresis in 56% of the patients included; other papers report that therapy is well tolerated with just 4–75% of adverse events.7,8
- ∘
Intravenous immunoglobulin: the European Federation of Neurological Societies indicated its administration in case of non-responders to first line therapies, at a dose of 2g/kg weight; a significant improvement of stiffness and a reduction of GAD autoantibodies were reported.8
- ∘
Monoclonal antibodies against B type receptor cd20+ lymphocyte cells, such as rituximab. The trials so far conducted emphasize good tolerance, long clinical remissions with improved spasms and muscle stiffness; however, the titers of circulating antibodies drop and the overall benefit is poor, probably on account of the antibody-mediated resistance mechanisms, the T-cells mediated immune activity rather than mediated by B-cells or auto-antibodies. Doses ranging from 350 to 375mg–m2 are administered every 7–14 days, or weekly, for 4 weeks.8 In GAD+ cases experiencing relapses, additional doses between months 6–8 of the initial infusion have been reported to be beneficial.1,5,8
- ∘
Pregnant patients with SPS should avoid benzodiazepine therapy during the first trimester because of the risk of teratogenicity; the use of baclofen is preferred. Benzodiazepines may be administered during the last trimesters, monitoring the risk of sedation, respiratory depression and fetal hypotony. Labor may be assisted with regional anesthesia and pain should be under control to avoid muscle spasms. There is no consensus in terms of the route of delivery – vaginal or cesarean section.11
ConclusionThe SPS is an infrequent condition with a broad range of differential diagnoses; comprehensive screening tests shall be performed, assessing the presence of antibodies to establish differences between a primary or secondary immune event associated with a paraneoplastic syndrome.
The treatment comprises symptomatic management and should include regimens with immunosuppressors and immunomodulators.
The clinical case herein discussed was considered to be relevant for publication on account of the low frequency of occurrence of this pathology, and in order to highlight the good clinical response and conspicuous quality of life improvement of the patient with plasmapheresis and monoclonal antibody treatment with rituximab.
FinancingNo financial contributions were received.
Conflict of interestsNone declared.
We would like to acknowledge Dr. Jorge Karim Assis, director of the Department of Research and Education, Clínica de Occidente S.A., and his team, for allowing us access to the institutional medical records in order to conduct this study.
Likewise, we acknowledge Prof. Adriana P. Henao Galindo, copy editor, for her contribution in reviewing this article.
Please cite this article as: González Trujillo F, Parra Cortes K, Barrios Arrazola G, Zapata Jaramillo JG. Síndrome de persona rígida, presentación de un caso clínico y actualidad en el tratamiento. Rev Colomb Reumatol. 2020;27:130–134.
