



Las vasculitis, patologías cuyo hallazgo principal es la afectación de las paredes de los vasos sanguíneos, se presentan de forma infrecuente en la población pediátrica. En general, en niños se clasifican de acuerdo con el consenso de la EULAR/PReS, y en adultos, según la Conferencia de Consenso de Chapel-Hill. Las vasculitis asociadas con ANCA (VAA) hacen parte de las vasculitis de pequeños vasos y están representadas por la granulomatosis con poliangeítis (GPA), la granulomatosis eosinofílica con poliangeítis (EGPA) y la poliangeítis microscópica (PAM), entre otras. A nivel renal, los hallazgos histopatológicos representativos son la glomerulonefritis focal necrotizante con media luna, inflamación intersticial variable, ausencia de complejos inmunes o pequeños depósitos de inmunoglobulinas. Clínicamente, las VAA pueden manifestarse con hematuria, proteinuria, hipertensión arterial o glomerulonefritis rápidamente progresiva. La GPA puede afectar de forma severa el riñón en el 75% de los casos, mientras que, en la PAM, el compromiso renal (75-90%) puede ser rápido y severo con posibilidad de requerir terapia de reemplazo renal en más de la mitad de los pacientes. Además, hasta el 25% de los casos puede tener hipertensión arterial, con una mortalidad a un año de 85%. En la EGPA, el compromiso renal suele ser leve. Se presentan 3 casos pediátricos de VAA con diferentes desenlaces renales, que incluyen necesidad de terapia de reemplazo renal con recuperación de función renal, trasplante renal y muerte, seguidas en una institución de IV nivel del suroccidente colombiano.
Vasculitis mainly affects the walls of the blood vessels, and is an uncommon disease in the paediatric population. In general, they are classified according to the EULAR / PReS consensus in children and in adults according to the Chapel-Hill consensus conference. ANCA-associated vasculitis (AAV) is part of small-vessel disease and is represented by granulomatosis with polyangiitis (GPA), eosinophilic granulomatosis with polyangiitis (EGPA), microscopic polyangiitis (MPA), and others. The representative renal histopathological findings are focal necrotising glomerulonephritis with crescents, variable interstitial inflammation, absence of immune complexes, or small deposits of immunoglobulins. Clinically, AAV can manifest with haematuria, proteinuria, high blood pressure, and/or rapidly progressive glomerulonephritis. GPA can severely affect the kidney in 75% of cases. Inn MPA renal involvement (75-90%) can be rapid and severe with the possibility of requiring renal replacement therapy in more than half of the patients. Furthermore, up to 25% of patients may have high blood pressure, and the mortality at one year can be up to 85%. In EGPA the renal involvement is usually mild. Three paediatric cases of AAV with different renal outcomes are presented, including the need for renal replacement therapy with the recovery of renal function, kidney transplantation, and death, followed in a fourth level of care institution in Colombia.
Las vasculitis pertenecen a un grupo heterogéneo de patologías, cuyo hallazgo principal es la afectación de las paredes de vasos sanguíneos de diferentes tamaños, lo cual es causa de daño en distintos órganos1. Su presentación en la edad pediátrica es poco frecuente. En general, los datos relacionados con la clínica, la clasificación y el manejo se extrapolan de población adulta.
El consenso de la European League against Rheumatism/Pediatric Rheumatology European Society (EULAR/PReS) clasifica las vasculitis en niños de acuerdo con el fenotipo clínico (afectación sistémica u órgano único), tamaño del vaso afectado (pequeño, mediano o grande) y la histopatología de los infiltrados2.
Las vasculitis de pequeños vasos afectan principalmente a vasos capilares y vénulas, aunque pueden comprometer a arterias y venas de mayor calibre3,4. Se dividen en dos entidades: a) las vasculitis granulomatosas, que incluyen patologías como la enfermedad granulomatosa con poliangeítis (GPA) (conocida anteriormente como granulomatosis de Wegener) o el síndrome de Churg-Strauss (conocido hoy en día como granulomatosis eosinofílica con poliangeítis [EGPA]); y b) las no granulomatosas, que incluyen la poliangeítis microscópica (PAM), diferente de la clasificación en adultos, la cual es dada según la Conferencia de Consenso de Chapel-Hill y divide a las vasculitis de pequeños vasos en vasculitis asociadas con anticuerpos anticitoplasma de neutrófilos (ANCA): GPA, PAM y EGPA, y vasculitis por inmunocomplejos, cuyo principal representante es la púrpura Henoch-Schönlein (PHS).
Los ANCA descritos son los anticuerpos para antígeno diana proteinasa 3 (PR3-ANCA) y los anticuerpos mieloperoxidasa (MPO-ANCA)5. Para fines epidemiológicos, la clasificación de Chapel-Hill ha resultado útil, y ha sido aplicada a población pediátrica. La determinación del reporte ANCA de los pacientes con vasculitis asociadas con ANCA es indispensable en el ejercicio clínico6.
En pediatría, las vasculitis asociadas con ANCA (VAA) tienen una presentación clínica heterogénea, con afectación frecuente del tracto respiratorio, el riñón, la piel y las articulaciones. Son infrecuentes en la infancia; se han reportado desde 0,5 casos/1.000.000 hasta 6,39/1.000.000, con pico de incidencia en la adolescencia. La mediana de edad en el momento del diagnóstico es aproximadamente de 11-14 años y se ha observado un predominio en mujeres (a diferencia de la población adulta, en la cual las VAA son más comunes en hombres)3,4.
Dentro de las vasculitis de pequeños vasos, la PHS es la más común en la edad pediátrica con una incidencia de aproximadamente 20/100.000 niños en países desarrollados y un pico de incidencia entre 4 y 6 años5,6. La tasa de mortalidad por VAA en la infancia es del 5-10%; los pacientes con EGPA, al parecer, tienen una mayor tasa de mortalidad estimada, la cual oscila alrededor del 15-18%. Diversos estudios han reportado una tasa de enfermedad renal en etapa terminal del 29-40% en PAM y del 10% en GPA5. Teniendo en cuenta la posibilidad de progresión a enfermedad renal terminal en estos pacientes, con requerimientos de terapia de reemplazo renal (TRR) y alta mortalidad asociada, es fundamental la caracterización de esta entidad en nuestra población infantil.
Los síntomas de las VAA incluyen síntomas constitucionales en el 50% de los pacientes (fiebre, fatiga, pérdida de peso y anorexia). El compromiso renal es común y puede ir desde hematuria, proteinuria o glomerulonefritis rápidamente progresiva, la cual se considera un predictor de mortalidad, especialmente en aquellos pacientes que debutan con tasas de filtración glomerular (TFG) inferiores a 50 miL/min/1,73 m2, en quienes se ha demostrado un riesgo de hasta el 50% para falla renal o muerte a los 5 años de seguimiento7.
Debido a que la sintomatología inicial en muchos casos es inespecífica, se puede retrasar significativamente el diagnóstico y, por ende, la demora en la instauración del manejo médico puede influir en el pronóstico renal5.
Se presentan 3 casos de pacientes femeninas en seguimiento por el servicio de nefrología pediátrica, cuyos casos fueron conocidos durante las hospitalizaciones en una institución de IV nivel del suroccidente colombiano. A todas se les confirmó la VAA por hallazgos histopatológicos en biopsias y el compromiso renal; solo una de ellas presentó ANCA positivos.
Reporte de casosPrimer caso: PAM ANCA negativoPaciente que en sus primeros años de vida presentó episodios de infección de vías urinarias a repetición, sin estudios de extensión. Posteriormente, a los 5 años debutó con cuadro de edemas, proteinuria e hipertensión arterial (HTA), por lo que fue hospitalizada con diagnóstico de síndrome nefrótico (SN) en otra institución. Paraclínicos durante la hospitalización pusieron en evidencia hematuria, proteinuria y elevación de azoados. A las 8 semanas se consideró SN corticorresistente por la ausencia de respuesta al manejo con esteroides y se practicó biopsia renal, la cual reportó glomerulonefritis proliferativa extracapilar con complemento sérico C3 y C4 normales.
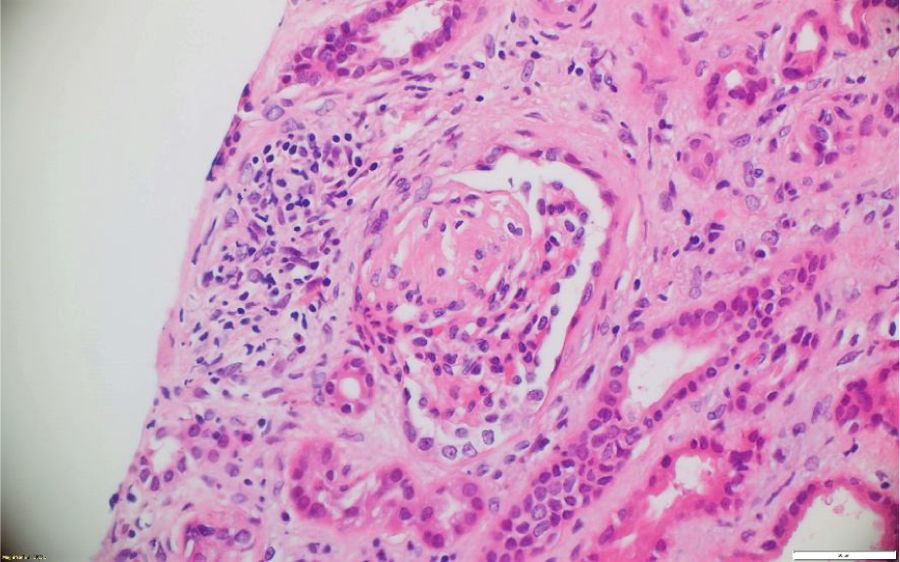
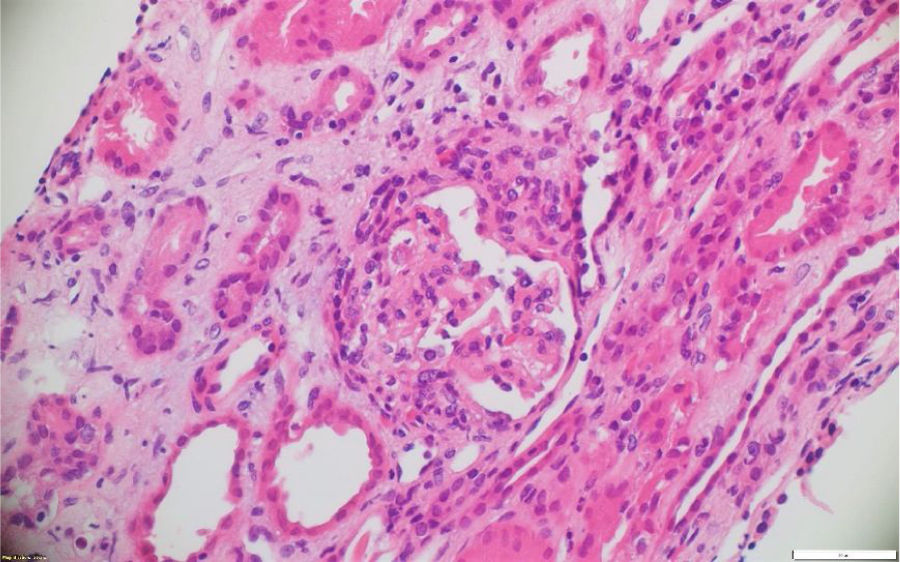
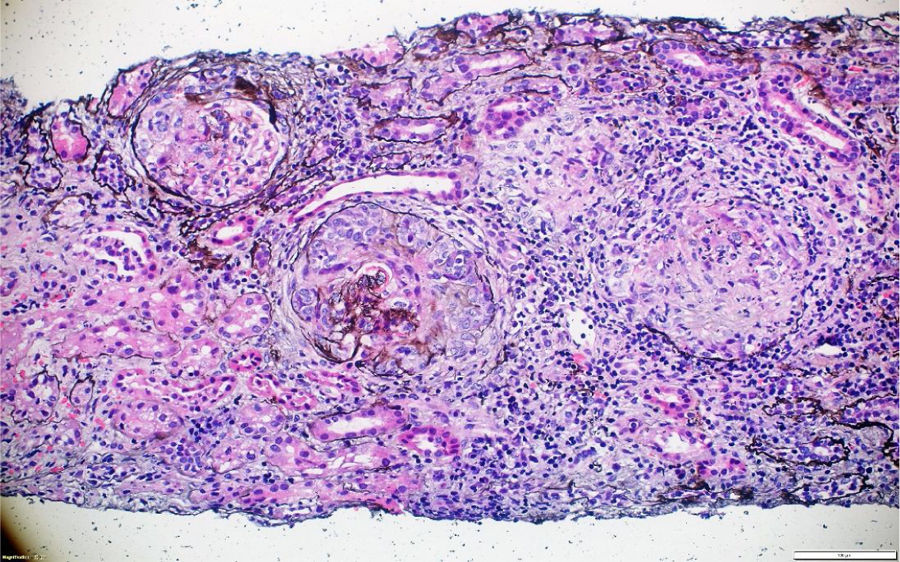
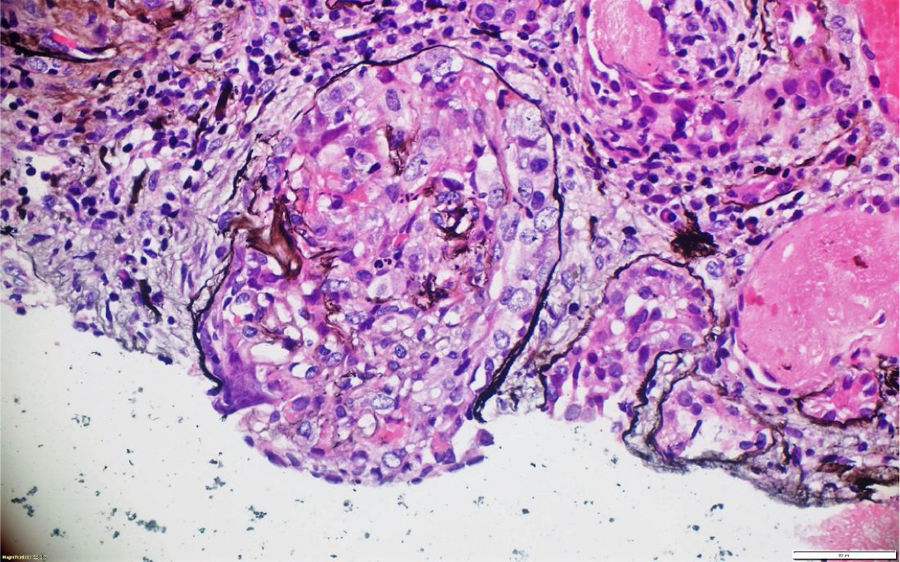
A pesar del manejo con pulsos de ciclofosfamida, la paciente presentó marcado deterioro clínico con requerimiento de diálisis peritoneal. Se realizó una segunda biopsia en nuestra institución, con evidencia de glomerulonefritis pauciinmune esclerosante con menos del 10% de semilunas fibrocelulares, fibrosis intersticial y atrofia tubular severa (figs. 1 y 2). Posteriormente, se tomaron niveles de MPO-ANCA y PR3-ANCA por inmunofluorescencia indirecta, los cuales fueron negativos.


Cuatro meses después de la segunda biopsia, y tras la confirmación del diagnóstico de vasculitis ANCA-negativo, la paciente presentó cuadro febril que se asoció con infección por influenza B y que requirió ventilación mecánica. Además, presentó anemización severa, derrame pleural, ascitis y hemorragia alveolar. El reporte de los ANCA fue negativo: PR3-ANCA = 1 U/mL (negativo < 5 U/mL) y MPO-ANCA 0,9 U/mL (negativo < 5 U/mL). También fueron negativos los ANA por inmunofluorescencia, en tanto que el reporte de anticuerpos anti-DNA (sustrato Crithdia luciliae) fue positivo con uno en 10 diluciones. La paciente fue valorada por reumatología pediátrica, que consideró una vasculitis ANCA-negativo con hemorragia alveolar difusa (síndrome riñón-pulmón), para lo cual recibió manejo con pulsos de esteroide, ciclofosfamida (750 mg/m2), plasmaféresis, hemodiálisis aguda y posterior diálisis peritoneal. Tuvo estabilización clínica y continuó manejo ambulatorio con azatioprina (3 mg/kg/día), prednisolona (1 mg/kg/día) y enalapril (0,25 mg/kg/día) durante 9 meses.
Después de 13 meses del diagnóstico de vasculitis, la paciente fue llevada a trasplante renal de donante fallecido y la evolución clínica fue satisfactoria. En la actualidad tiene 11 años y asiste a seguimiento mensual intrainstitucional con tasa de filtración glomerular de 80 mL/min/1,73 m2, recibe como inmunosupresión tacrolimus (dosis de 0,15 mg/kg/día) y mofetilmicofenolato (dosis de 600 mg/m2/día).
Segundo caso: PAM ANCA positivoPaciente de 13 años con cuadro clínico de 2 meses de evolución de palidez mucocutánea progresiva asociada con síndrome febril de 20 días de evolución y astenia. Al examen físico se evidenció presencia de nódulos supurativos en cuero cabelludo y eritema faríngeo. También presentó artralgia de rodilla izquierda con lesiones vasculíticas, lesiones tipo pápulas violáceas que no desaparecían con la digitopresión.
Paraclínicos de ingreso evidenciaron anemia (hemoglobina de 8,9 g/dL), hematuria y proteinuria (75 mg/dL) en muestra espontánea de orina, elevación de azoados (BUN = 45 mg/dL, creatinina sérica = 2,9 mg/dL), complemento sérico C3 y C4 normales, anticuerpos antinucleares y anticuerpos antimembrana basal glomerular negativos, PR3-ANCA negativo < 3,5 U/mL (negativo < 5 U/mL) y MPO-ANCA positivo 72 U/mL (negativo < 5 U/mL). Al ingreso se sospechó glomerulonefritis postestreptocócica complicada versus nefritis lúpica. Ante el aumento progresivo de los azoados y del deterioro clínico de la función renal, se requirió inicio de hemodiálisis y practicar biopsia renal.
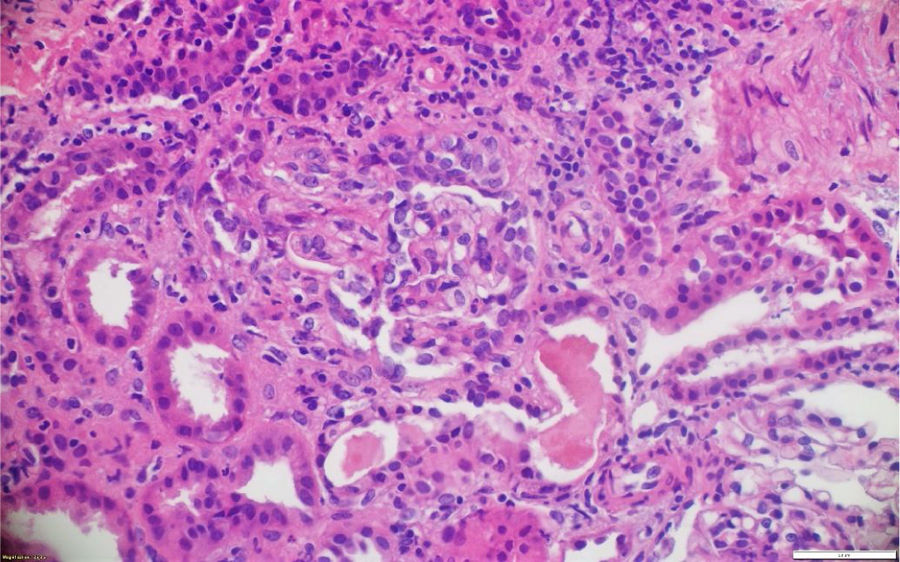
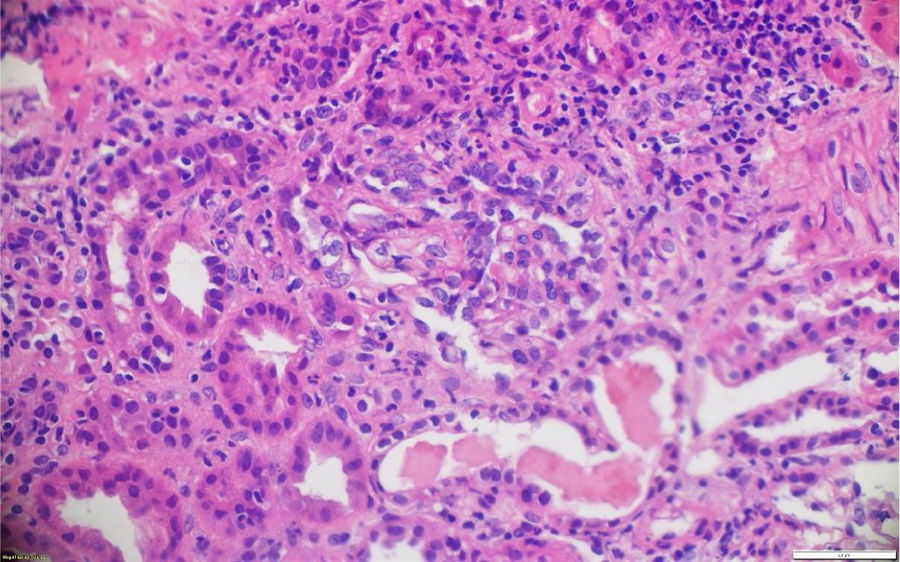
La patología renal mostró proliferación crescéntica activa de origen celular, con actividad alta, necrosis fibrinoide extensa, infiltración leucocitaria y evidencia de infiltrado linfoplasmocitario en el intersticio, asociado a 10% con fibrosis. No había evidencia de depósitos de complejos inmunes (figs. 3 y 4).


La paciente continuó con deterioro de la función renal. En conjunto con reumatología pediátrica y nefrología pediátrica, se decidió iniciar manejo con plasmaféresis, ciclofosfamida y rituximab. De ciclofosfamida recibió un total de 6 dosis de 10 mg/kg/dosis: cada 2 semanas las primeras 3 dosis y posteriormente cada 3 semanas las otras 2 dosis; y de rituximab 4 dosis: una dosis semanal de 375 mg/m2/dosis. La azatioprina fue suspendida por leucopenia y se continuó con mofetilmicofenolato (MMF) (1.100 mgr/m2/día). La paciente presentó adecuada evolución clínica y mejoría de la función renal, por lo que se suspendió hemodiálisis a los 3 meses del inicio del manejo inmunosupresor.
En la actualidad la paciente tiene 18 años, se encuentra en seguimiento por reumatología con diagnóstico de PAM, permanece asintomática, tiene creatinina sérica de 1 mg/dL y recibe manejo con MMF (1.100 mgr/m2/día).
Tercer caso: GPA ANCA negativoEl tercer caso se presentó en una paciente de siete años que debutó con cuadro clínico de dos años de evolución consistente en episodios de infección respiratoria a repetición y estridor, manejado ambulatoriamente con esteroide en múltiples oportunidades. Tres meses antes del ingreso presentó hiporexia, decaimiento y hematuria macroscópica. Posteriormente desarrolló edema generalizado. Al examen físico presentó masa intraorbitaria izquierda, proveniente de la glándula lagrimal y las lesiones descritas con anterioridad.
Durante la evolución presentó HTA, proteinuria en rango nefrótico, C3 y C4 normales y anticuerpos antinucleares y anti-DNA negativos. Se realizaron ANCA por medio de IFI, los cuales reportaron PR3-ANCA positivos (1:160 diluciones) y MPO-ANCA negativos. Sin embargo, se realizaron MPO-ANCA y PR3-ANCA por ELISA, los cuales fueron negativos (PR3-ANCA < 0,2 U/mL [negativo < 5 U/mL] y MPO-ANCA 0,8 U/mL [negativo < 5 U/mL]).
Se consideró que la paciente cursaba con GPA y se inició inmunosupresión con ciclofosfamida (en total 6 dosis de 10 mg/kg/dosis, cada 2 semanas las primeras 3 dosis y posteriormente cada tres semanas las otras 2 dosis) y rituximab (4 dosis semanales de 375 mg/m2/dosis). Sin embargo, a pesar del manejo instaurado, la paciente presentó un rápido deterioro de la función renal y requirió diálisis peritoneal.
A los 10 años presentó múltiples complicaciones derivadas de granulomas en piel, mucosas (incluyendo esófago) tejidos blandos y óseos.
La biopsia de piel de la lesión ulcerativa en muslo izquierdo tuvo un reporte de patología que mostró abundante infiltrado inflamatorio mixto de predominio polimorfonuclear supurativo, con áreas de ulceración extensa, tejido de granulación con proliferación de vasos y abundantes histiocitos. Adicionalmente, la biopsia renal reportó presencia de medialunas fibrocelulares en más del 50% de los glomérulos, proliferación de células mesangiales e inmunofluorescencia negativa para complejos inmunes (figs. 5 y 6).


La paciente presentó un gran deterioro generalizado, con peritonitis esclerosante como complicación mayor, lo cual obligó a cambio de TRR de diálisis peritoneal a hemodiálisis. Falleció en el contexto de sepsis por enfermedad fúngica invasora.
DiscusiónEl término vasculitis, como ya se mencionó, se usa para identificar a un grupo heterogéneo de patologías cuyo hallazgo principal es la afectación de las paredes de los vasos sanguíneos1.
Específicamente para el diagnóstico de la granulomatosis de Wegener, el consenso EULAR/PReS describió la necesidad de positividad de al menos 3 criterios (alteraciones en el parcial de orina tipo hematuria/proteinuria significativa, presencia de inflamación granulomatosa en biopsia, inflamación del seno nasal, estenosis subglótica, traqueal o endobronquial, radiografía de tórax o tomografía anormal y tinción ANCA positiva)2.
En contraste, en los adultos las vasculitis se clasifican de acuerdo con el consenso de Chapel-Hill, en el que a su vez se agrupan según el tamaño del vaso comprometido, en grandes, medianos y pequeños vasos, vasculitis de vaso variable, de órgano único, asociadas con enfermedad sistémica o asociadas con etiología probable.
Para las vasculitis de pequeños vasos aparecen los términos de granulomatosis con poliangeítis (granulomatosis de Wegener), granulomatosis eosinofílica con poliangeítis (síndrome de Churg-Strauss) y poliangeítis microscópica, todas bajo una misma denominación de VAA8. Las VAA ANCA negativo son un subgrupo con características clínicas e histológicas de VAA, pero con reporte de anticuerpos negativos; la literatura describe un aparente compromiso renal limitado o sistémico menos severo en estos pacientes7.
El compromiso renal en el contexto de VAA puede manifestarse como hematuria, proteinuria, HTA o glomerulonefritis rápidamente progresiva. La GPA se caracteriza por inflamación granulomatosa y necrosis. Aunque tiene predilección por el tracto respiratorio (80%), el riñón también se puede comprometer de forma severa hasta en el 75% de los casos2,9. Se asocia con MPO-ANCA en un 20%, PR3-ANCA en un 75% y ANCA negativo en un 5%7,10.
La tercera paciente también tuvo debut con hematuria, sumado a proteinuria e HTA, reporte de MPO-ANCA y PR3-ANCA por ELISA negativos, biopsia renal con predominio de medialunas y falla terapéutica con necesidad de diálisis peritoneal. En Colombia, hay un caso pediátrico descrito de debut clínico con hemorragia alveolar, hematuria, oliguria, requerimiento de terapia de reemplazo renal y PR3-ANCA títulos altos, sumados a hallazgos en biopsia renal de compromiso por glomerulonefritis necrosante focal y segmentaria con proliferación extracelular y presencia de medialunas en más del 70% de los glomérulos, sin recuperación de la función renal, pese al manejo con esteroides, ciclofosfamida y plasmaféresis11. Otras series colombianas reportan manejo con rituximab a pacientes pediátricos con GPA sin compromiso severo renal12.
Con respecto al reporte de ANCA del tercer caso por IFI positivo y por ELISA negativo, la literatura describe que las técnicas para medir la positividad de ANCA son ELISA e inmunofluorescencia (IFI). Si las dos pruebas muestran positividad para ANCA, el valor predictivo positivo (VPP) está descrito hasta en el 88% de los casos. El test por IFI tiene un grado de subjetividad, pues la prueba se basa en la valoración visual del patrón de inmunofluorescencia, por lo que el resultado puede depender de la experticia del lector. Con respecto a la técnica por ELISA, esta se asocia con mayores porcentajes de especificidad (E) y VPP, comparada con la inmunofluorescencia (E 82% y VPP 63% vs. E 18% y VPP 13%, respectivamente)13,14. En población adulta generalmente se usan de forma combinada IFI y ELISA15.
La PAM es menos frecuente en niños y a diferencia de la GPA no presenta inflamación granulomatosa9. Se asocia con MPO-ANCA en un 60%, PR3-ANCA en un 30% y ANCA negativos en un 10% de los casos7. En pediatría, el compromiso cutáneo tipo púrpura es común como presentación, y también es frecuente el compromiso neurológico (21-86%) y pulmonar (44%). La afectación renal (75-90%) puede ser rápida y severa con posibilidad de requerir TRR en más de la mitad de los pacientes. Hasta el 25% de los pacientes puede tener HTA y la mortalidad a un año es hasta del 85%16,17.
Las pacientes del primer y del segundo caso debutaron de forma diferente desde el punto de vista renal: la primera de ellas como SN, MPO-ANCA y PR3-ANCA negativos e historia de manejo con esteroide, ciclofosfamida y plasmaféresis. Y la segunda con hematuria y proteinuria no severa, MPO-ANCA positivo e historia de manejo con esteroide, ciclofosfamida, plasmaféresis y rituximab. Sin embargo, ambas tuvieron deterioro con necesidad de TRR, una de ellas sin recuperación y requerimiento de trasplante renal y la otra con recuperación de función renal 3 meses después del inicio de la hemodiálisis.
La EGPA, enfermedad que genera inflamación granulomatosa rica en eosinófilos, es aún menos común en niños y se ha relacionado con asma y atopia1. El compromiso renal es usualmente leve y se asocia con MPO-ANCA en un 45%, PR3-ANCA en un 5% y ANCA negativo en un 50%7,18. Es la vasculitis que mayor proporción de ANCA negativo presenta si no hay afectación renal y en nuestro servicio de nefrología pediátrica no hemos tenido casos representativos.
El diagnóstico del compromiso renal se establece mediante biopsia. Las VAA causan glomerulonefritis que se caracteriza por infiltrados en las paredes de los vasos, de predominio de polimorfonucleares (neutrófilos, eosinófilos)1. Los hallazgos histopatológicos tienen buena correlación clínica, el patrón histopatológico distintivo es la presencia de necrosis de las asas de los capilares, proliferación extracapilar con formación de crecientes o medialunas, infiltrado intersticial y periglomerular, arteritis necrotizante y ausencia o pauciinmunidad de complejos inmunes1,19.
Berden et al. idearon un sistema de clasificación histopatológica para confirmar las glomerulonefritis asociada con ANCA, denominada focal (más del 50% de glomérulos normales), crecientica (más del 50% de los glomérulos con medialunas), mixta (menos del 50% de los glomérulos normales, menos del 50% con medialunas y más del 50% con esclerosis global) y esclerótica (más del 50% de los glomérulos globalmente esclerosados)20. Para pacientes clasificados como focal, la sobrevida renal a un año es del 93%, 84% para pacientes clasificados como crecientica, 69% para pacientes clasificados como mixta y 50% para los clasificados como esclerótica.
Los casos aquí reportados tuvieron variabilidad en el reporte histológico: el clasificado como GPA tuvo medialunas en más del 50% de los glomérulos, en concordancia con pérdida de sobrevida renal y necesidad de diálisis. Los clasificados como MPA presentaron hallazgos histológicos diferentes. El PAM ANCA negativo tuvo como desenlace deterioro de la función renal y trasplante renal. En dichos casos predominó la fibrosis intersticial y la atrofia tubular severa, mientras que en el PAM ANCA positivo lo hizo la necrosis fibrinoide, la cual tuvo recuperación de la función renal posteriormente a la recaída renal.
En general, el pronóstico renal se relaciona con múltiples hallazgos, algunos histológicos predominantes como el número de glomérulos normales, el grado de tubulitis e inflamación intersticial y la presencia de esclerosis o crecientes/medialunas en la biopsia renal. Estudios de seguimiento describen posibilidad de desarrollo de enfermedad renal terminal hasta en el 40% de los pacientes con MPA y 10% en GPA5.
ConclusionesLas VAA son un grupo de enfermedades multisistémicas poco frecuentes en la edad pediátrica, cuyos síntomas generales al inicio dificultan y hacen inespecífico el diagnóstico en etapas tempranas. Se destaca en los 3 casos presentados la variabilidad clínica y los diferentes desenlaces renales de una misma patología, que incluyen recuperación de la función renal, terapia de reemplazo renal, trasplante y muerte.
Consentimiento informadoLos autores declaran que se obtuvieron los consentimientos informados firmados de los pacientes o sus progenitores y también la aprobación del comite ético de la institución.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
