





Vogt Koyanagi Harada disease affects several parts of the body, such as eyes, meninges, ears, and skin. The progressive course of the disease can lead to blindness and deafness. The case is presented of a Hispanic woman (mixed-race) with visual alterations, headache, tinnitus, hearing loss, and posterior uveitis with serous detachments of the retina in both eyes, as well as lymphocytic meningitis. The aim of the present study is to review the literature, the diagnostic strategies, and the appropriate treatment, as well as to update the immunogenetic pathogenesis of the disease.
La enfermedad de Vogt Koyanagi Harada compromete múltiples órganos tales como ojos, meninges, oídos y piel. El curso progresivo de la enfermedad puede llevar a ceguera y cofosis. Se describe un caso de esta enfermedad en mujer hispana (mestiza) con alteraciones visuales, cefalalgia, tinnitus e hipoacusia a quien se le encuentra uveítis posterior con desprendimientos serosos de retina en ambos ojos y meningitis linfocitaria. El objetivo del presente estudio es, mediante una revisión de la literatura, actualizar la patogénesis inmunogenética, conocer las estrategias diagnósticas y el tratamiento apropiado.
A 34-year old female patient, working as oral hygienist, who attended the external consultation of ophthalmology for a clinical picture of 6 weeks of evolution consisting in seeing the straight lines of the road signs distorted and bright an at night not perceiving with certainty the direction of moving cars; in addition, blurred vision and myodesopsies. Associated with the previous picture, the patient reports episodes of 3 weeks of evolution of headache, persistent tinnitus, hypoacusis and visual deterioration with compromise of color vision. She also presented isolated episodes of "forgetfulness for seconds" that caused her short and transient blockages in the course of thinking and language. As a relevant antecedent, she referred a family history of glaucoma.
The ophthalmological exam showed convergence insufficiency and exotropia. Visual acuity 20/20 in both eyes (OU), however, she manifested distortion of the visual object with central vision in OU and alteration in the color vision, greater in the right eye (OD). Bulbar conjunctival hyperemia in OU, clear corneas without keratic precipitates, Tyndall (−), iris and pupils of healthy appearance, normal photomotor and consensual reflexes in OU, without afferent pupillary defect were identified in the exam. Intraocular pressure of 12 mmHg in OU. Fundus of the eye with clear media, optic disc-excavation ratio (cup) 0.6/0.6, the thickness of the neuroretinal rim around the optic papillae (inferior, superior, nasal and temporal) is preserved, bayonet vessels, slight nasalization of the vessels, artery-vein ratio 2/3, macules with decreased brightness and presence of bilateral macular folds, greater in the OD macule with apparent epimacular membrane (pucker). The initial diagnostic impression of ophthalmology was glaucoma, epiretinal membrane and posterior uveitis. Retinal fluorescein angiography, fundus photographs, optical coherent tomography of optic discs and macules and 24-2 perimetry of OU were requested. Due to the headache and cognitive alterations, the patient was referred to neurology where, in addition to the ophthalmological findings described, they found her nuchal rigidity. Based on the above, a syndromic diagnosis was established:
- 1
Cranial nerves syndrome: bilateral ii cranial nerve due to maculoretinal involvement and viii cranial nerve by bilateral auditory commitment.
- 2
Menigoencephalic syndrome, etiology to be studied.
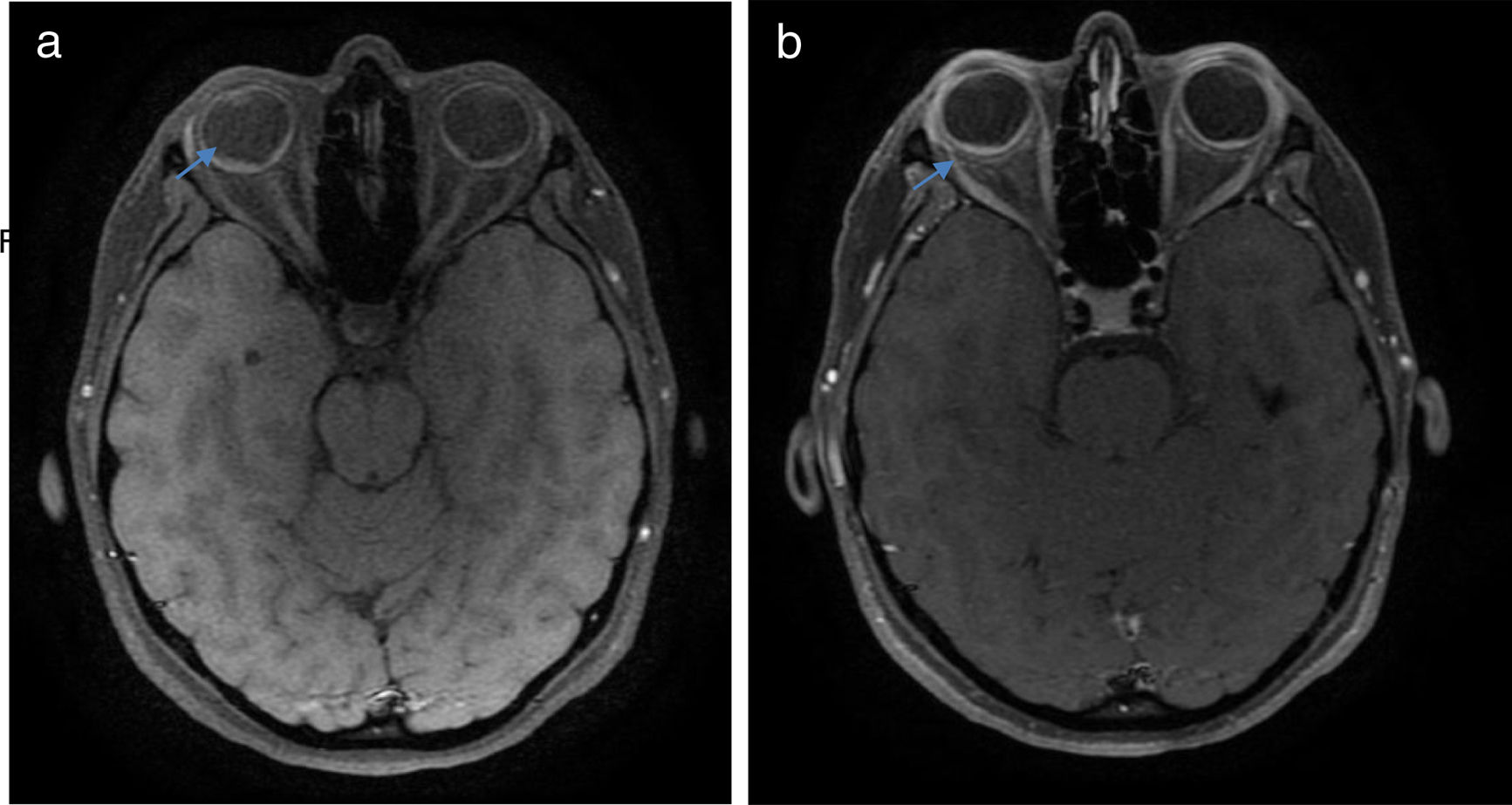
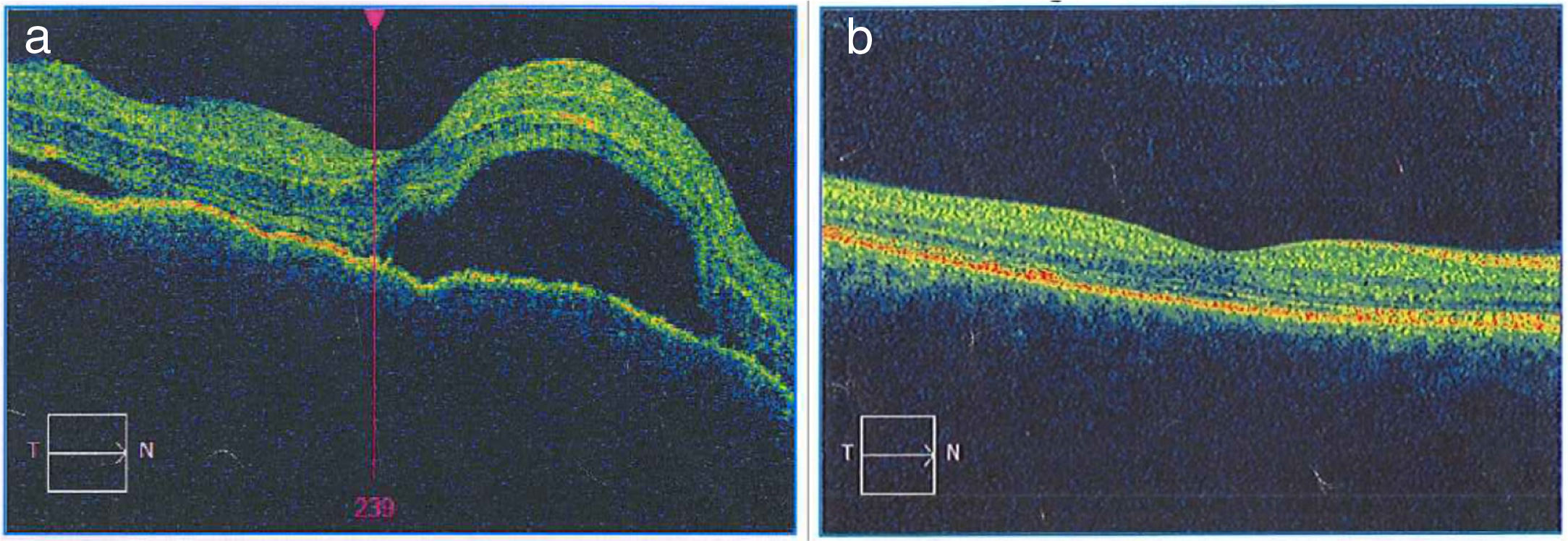
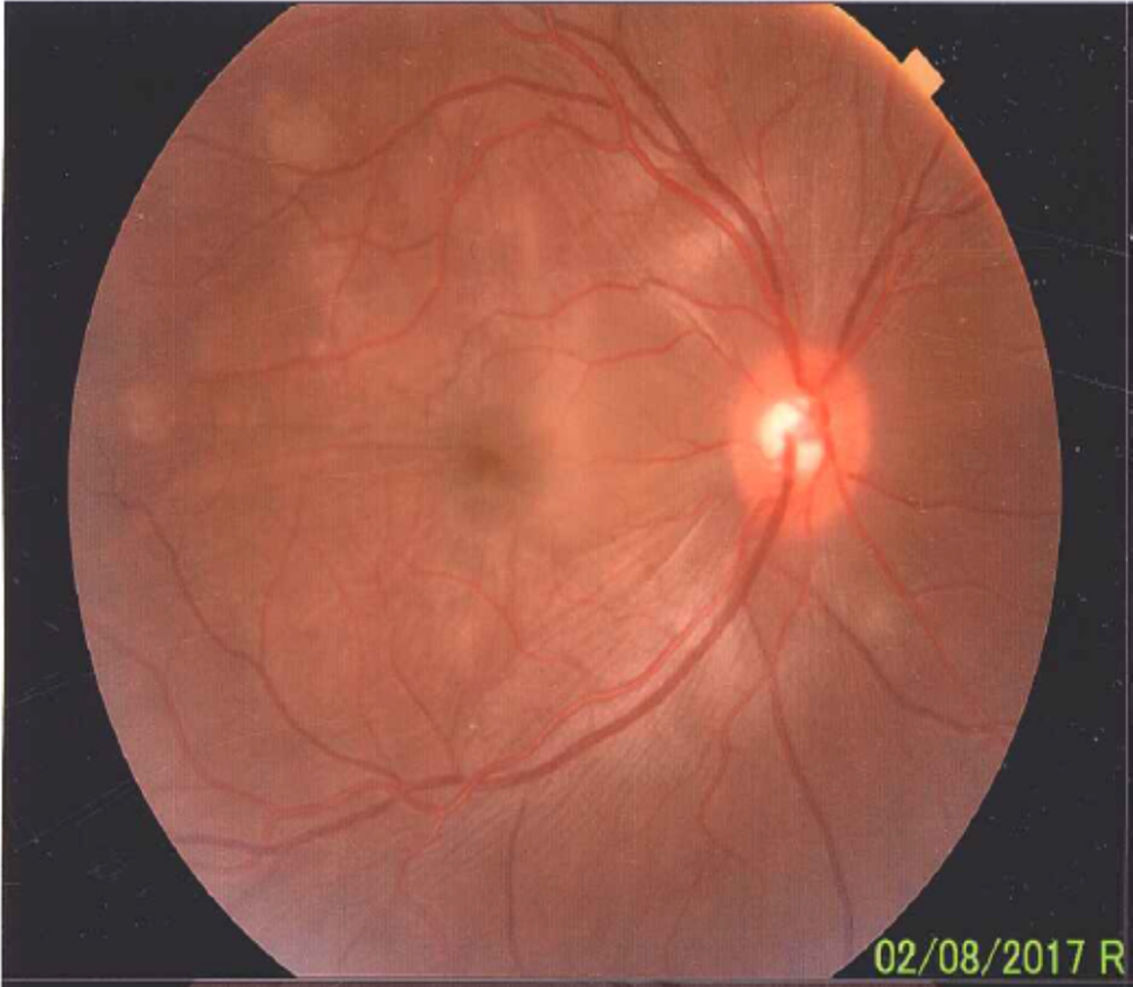
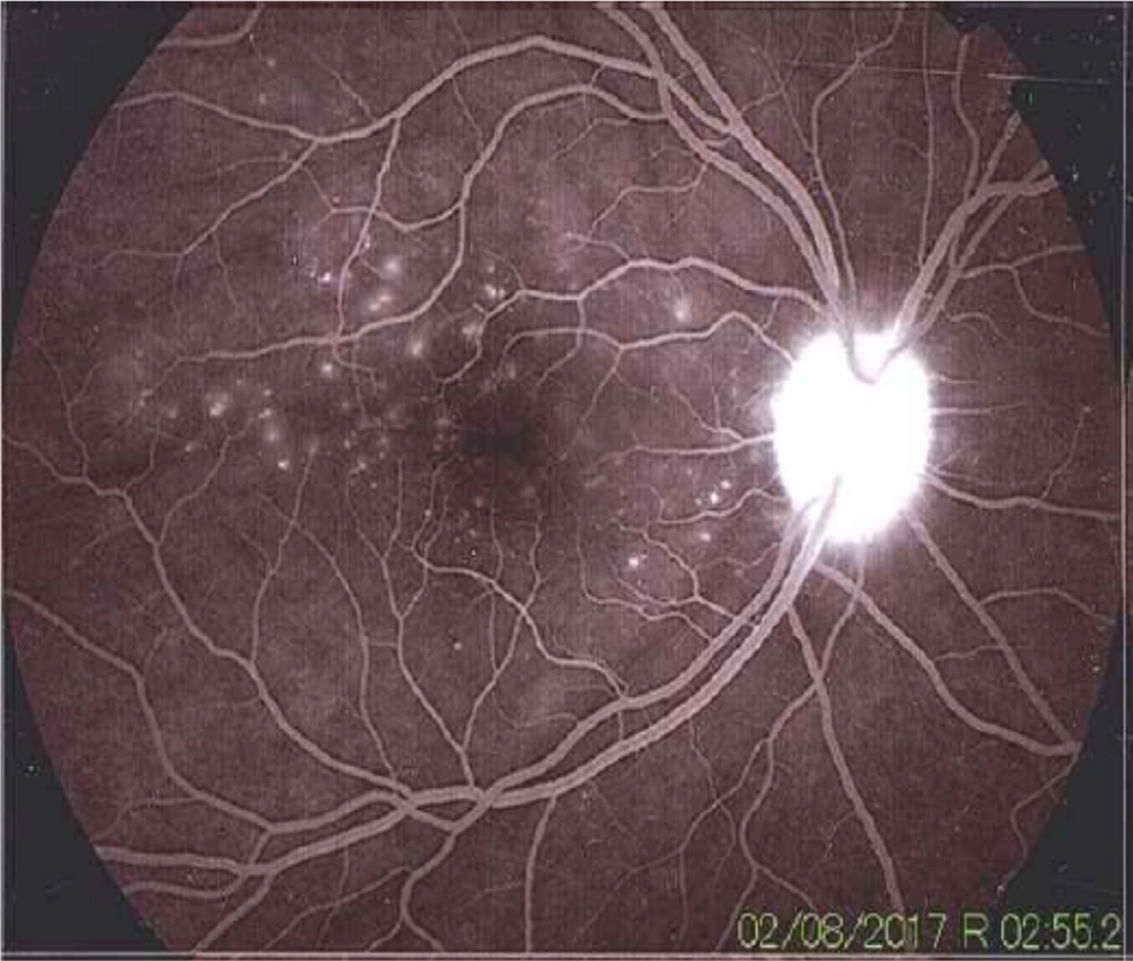
Once the syndromic diagnosis was established, the patient was hospitalized for study and treatment. Paraclinical tests were requested with results reported within normal parameters (Table 1). Magnetic resonance imaging (MRI) of the brain, simple and with contrast, without parenchymal alterations and without meningeal enhancement. In the orbits, the MRI showed an increase in the thickness of the choroid in both eyeballs with intact scleras, findings compatible with inflammatory ocular process (Fig. 1). The optic coherence tomography (OCT) of the macule (Fig. 2a) showed choroidal thickening and serous retinal detachments. The photography of the eye fundus (Fig. 3) and the digital fluorescein angiography (Fig. 4) demonstrated uveoretinal lesions with compromise of the pigment epithelium and serous retinal detachments. The cerebrospinal fluid (CSF) study obtained through a lumbar puncture (LP) reported lymphocytic pleocytosis. The audiometry recorded bilateral sensorineural hearing loss. A definitive diagnosis of uveomeningoencephalic syndrome due to Vogt Koyanagi Harada (VKH) disease is established based on the history, the clinical findings, laboratory results and images.
Laboratory tests.
| Hemoglobin: 14.50 g/dl |
| Hematocrit: 44 % |
| Normal leukocyte count |
| Erythrocyte sedimentation rate (ESR) 15.00 mm/hour (normal up to 25 mm/hour) |
| Glucose: 80 mg/dl |
| Antinuclear antibodies (ANA) (IIF method): 1/80 |
| TSH: 2.75 mU/ml (0.27–4.20) |
| Free tyroxine T4: 0.89 ng/dl |
| Creatinine: 0.80 mg/dl |
| Blood urea nitrogen (BUN): 20.3 mg/dl |
| C reactive protein: 3.00 mg/l (normal 0−5 mg/l) |
| VDRL serology: non-reactive |
| Sodium: 138 meq/l |
| Potassium 4.01 meq/l |
| ENA: negative |
| Pyruvic transaminase (GPT-ALT): 14 U/L |
| Oxaloacetic transaminase (GOT-AST): 18 U/L |
| Urinalysis: normal |
| Cytomegalovirus IgG antibodies (electrochemiluminiscence method): 348.90 U/ml |
| Cytomegalovirus IgM antibodies (ELFA method): 0.22 negative index less than 0.7 |
| Epstein Barr Virus IgG (ELFA method): 1.860 |
| Epstein Barr Virus IgM (ELFA method): 0.010 negative index lower than or equal to 0.11 |
 and after (b) contrast, with emphasis on the orbits, which show increased choroidal thickness, with diffuse enhancement after contrast (arrows).")
 The macular OCTdiagnostic image of the OD allows to observe the neuroepithelium with an irregular surface, retinal folds in the internal limiting membrane, loss of foveal depression with a generalized increase in macular thickness. Multiple serous detachments of the neuroepithelium with accumulation of subretinal fluid are also observed, images that confirm the Vogt Koyanagi Harada disease. b) Resolution of the retinal detachments and the RPE-Bruch’s membrane-choriocapillaris complex without alterations can be clearly seen in the macular OCT of the OD taken 6 months after the initiation of treatment. The choroid appears with normal pattern and thickness.")
a) The macular OCTdiagnostic image of the OD allows to observe the neuroepithelium with an irregular surface, retinal folds in the internal limiting membrane, loss of foveal depression with a generalized increase in macular thickness. Multiple serous detachments of the neuroepithelium with accumulation of subretinal fluid are also observed, images that confirm the Vogt Koyanagi Harada disease. b) Resolution of the retinal detachments and the RPE-Bruch’s membrane-choriocapillaris complex without alterations can be clearly seen in the macular OCT of the OD taken 6 months after the initiation of treatment. The choroid appears with normal pattern and thickness.

A preserved artery-vein ratio, disc with well-defined borders, yellow-pink, excavation 0.4, normal vascular emergence and distribution can be observed in the eye fundus. The posterior pole, macular area and peridiscal region show folds in the internal limiting membrane and diffuse defects of the yellow in patches.

The angiography showed adequate circulatory dynamics of the medium with hyperfluorescence of the disc. Multiple hyperfluorescent defects due to dye leakage, suggestive of serous retinal detachments can be observed in the photo of the posterior pole. Punctiform hyperfluorescent defects are also shown in the macular area.
Treatment was started with boluses of methylprednisolone (1 g/day for 4 days), followed by a scheme of prednisolone (60 mg/day and 10 days), then methotrexate (20 mg/week) was added. The patient showed rapid clinical improvement during the first week of treatment: the headache, the tinnitus and the meningism subsided. The patient was assessed by a retinologist, and given the presence of macular edema and cystoid retinal edema, 40 days after initiation of treatment, it was decided the intravitreal application of ranibizumab in the OD: one dose and corticosteroid eye drops in the OD for 7 days. Periodic controls demonstrated progressive improvement, total recovery of visual acuity and chromatic vision, with preservation of her visual function after 12 months of follow-up. After the sixth month of treatment, a slow and progressive reduction of the oral corticosteroid was started. At the eighth month of treatment she presented left intercostal neuritis due to varicella zoster virus that responded well to treatment with valciclovir. The convergence insufficiency and exophoria persisted. The evaluation of the treatment has been accompanied by OCT images (Fig. 2b) that demonstrate disappearance of retinal and macular edema and well applied retina, with reversal of retinal serous detachments at 6 months of treatment.
MethodologyThis paper exposes the case of a patient studied and treated by the Colsanitas healthcare provider network in Bogotá and associates it with a literature review. The development of the literature review and search started with the formulation of the qualitative research question taking as a guide, in its structuring, the PICO methodology, an acronym where «P» is the population under study, «I» makes reference to a form of medical intervention, C is the «comparator» with another form of intervention and O comes from «outcome».
In middle-aged individuals residing in Colombia who consult for presenting ocular alterations, myodesopsia, dyschromatopsia, headache, meningism and auditory-vestibular symptoms (population, P), is it possible to establish the clinical diagnosis of VKH disease (intervention, I), or it is only possible to make the diagnosis of VKH through the practice of paraclinical studies requested from a syndromic diagnosis (comparator, C), in order to start treatment timely and safely (outcome, O)?
This review finds justification in the need to obtain updated knowledge of VKH, a rare and complex disease, which is very occasionally reported in Colombia. The purpose is to raise awareness about the importance of rapid diagnosis, timely treatment with specialized follow-up, aimed at avoiding the severe complications that accompany it. This work is a narrative review of the recent literature on VKH disease. The articles were selected by searching in the databases: Pubmed and Google Scholar. The search keywords or descriptors used were in English: uveomeningoencephalitic syndrome and Harada; in Spanish: síndrome uveomeningoencefálico y Harada.
For the correct use of the terminology, the 2017 edition of the Health Science Descriptors was consulted in the Website:
The research of available evidence was performed using the search descriptors. The period between January 2010 and December 2017 was decided as search interval. 6103 potentially relevant articles were identified in English: 353 works in Pubmed and 5750 in Google Scholar. The review of the articles was carried out between the months of June and December 2017 by 2 independent researchers.
For the selection of the articles it was taken into account that they were published in the indicated period, the methodological quality, participation of expert authors, as well as the usefulness and relevance of the contents regarding the research question. 35 articles that met the requirements were finally selected for the review, 17 from Pubmed and 18 from Google Scholar. In order to limit the selection bias, strategies such as the orderly search of the articles supplied by the databases consulted and the inclusion of works obtained from the gray literature were implemented. One of the articles selected for the review was published prior to the search period, but it is considered a mandatory reference since it contains the review of diagnostic criteria for VKH disease that are still in force.
Literature reviewBackgroundVKH disease is a multisystemic disorder characterized by ocular inflammation with neurological, hearing and dermatological symptoms and signs. The disease was initially described in 1906 by the Swiss ophthalmologist Alfred Vogt, who reported the case of a patient with iridocyclitis and poliosis. In a review article, published in 1929, Yoshizo Koyanagi described 16 cases illustrating the course of the disease. Einosuke Harada, internist and ophthalmologist, in addition to extraocular findings describes acute posterior choroiditis with exudative retinal detachments and pleocytosis in the CSF.1 Harada was the one who managed to make the full description of what is known today as VKH disease.
EpidemiologyVKH disease has a variable incidence, more frequent in people from East Asia, Hindus, Hispanics, Mediterranean and Native Americans; it is infrequent in Caucasians and usually affects patients between the ages of 20 and 50 years, although it also occurs in children.1 Studies indicate that women are more committed than men, with a 2:1 ratio, except some studies from Japan and China, which showed no differences in prevalence by gender.2
Clinical manifestationsThe clinical course of the disease has been described by the international committee of experts in VKH, convened by the American Uveitis Society, in an article published in 2001,3 which contemplates the 4 classic chronological phases: prodromal, acute uveitic, convalescent and chronic recurrent.
Prodromal phasePatients who develop the prodromal phase simulate a systemic viral picture with clear neurological manifestations consisting of headache, orbital pain, nausea, hypoacusis or tinnitus, nuchal and back rigidity (meningism) and scalp hyperesthesia. These clinical manifestations range from 3 to 15 days before the acute uveitic phase.1 In this phase, cerebral focal signs such as confusion, aphasia and hemiparesis are rarely present.
Meningism is the most common extraocular clinical sign, present in 49–67 % of patients with VKH.4 Lymphocytic pleocytosis in the CSF is found in more than 80 % of patients and it can persist for up to 8 weeks.3,5 Other changes in the CSF include the presence of melanin-laden macrophages specific for VKH, increase in proteins and opening pressure.2 The CSF analysis is an important test for the early diagnosis of VKH, particularly in patients with headache, clear meningeal signs and few ocular signs6; however, ophthalmologists experts in uveitis, who work in geographic areas endemic for VKH and in specialized reference centers, affirm that they rarely need the study of CSF for the diagnosis of VKH disease and suggest that LP should not be a routine procedure, given that the clinical findings and the advanced imaging techniques in ophthalmology, such as fluorescein angiography and OCT, confirm the clinical diagnosis.5,6
Sensorineural hearing loss, tinnitus, aural fullness and vertigo usually appear in the prodromal phase and can be concomitant with active uveitis.7 Subjective sensorineural hearing loss was demonstrated in approximately 40 % of patients, while tinnitus was similarly reported in 38 %.8,9 Vestibular symptoms such as vertigo and dizziness occurred less frequently, and are described in up to 25 % of patients with VKH, that is, in 1 out of every 4.10 The pattern of hearing loss is bilateral and mild, mainly in the high frequency range9,10; however, cases of sudden and severe bilateral deafness are reported.11 Remarkably, the hearing loss detected by audiometry is close to 90 % of cases, significantly greater than the subjective hearing loss (27 %).9 These audiological findings have allowed to recommend audiometry as part of the evaluation and control of the disease.
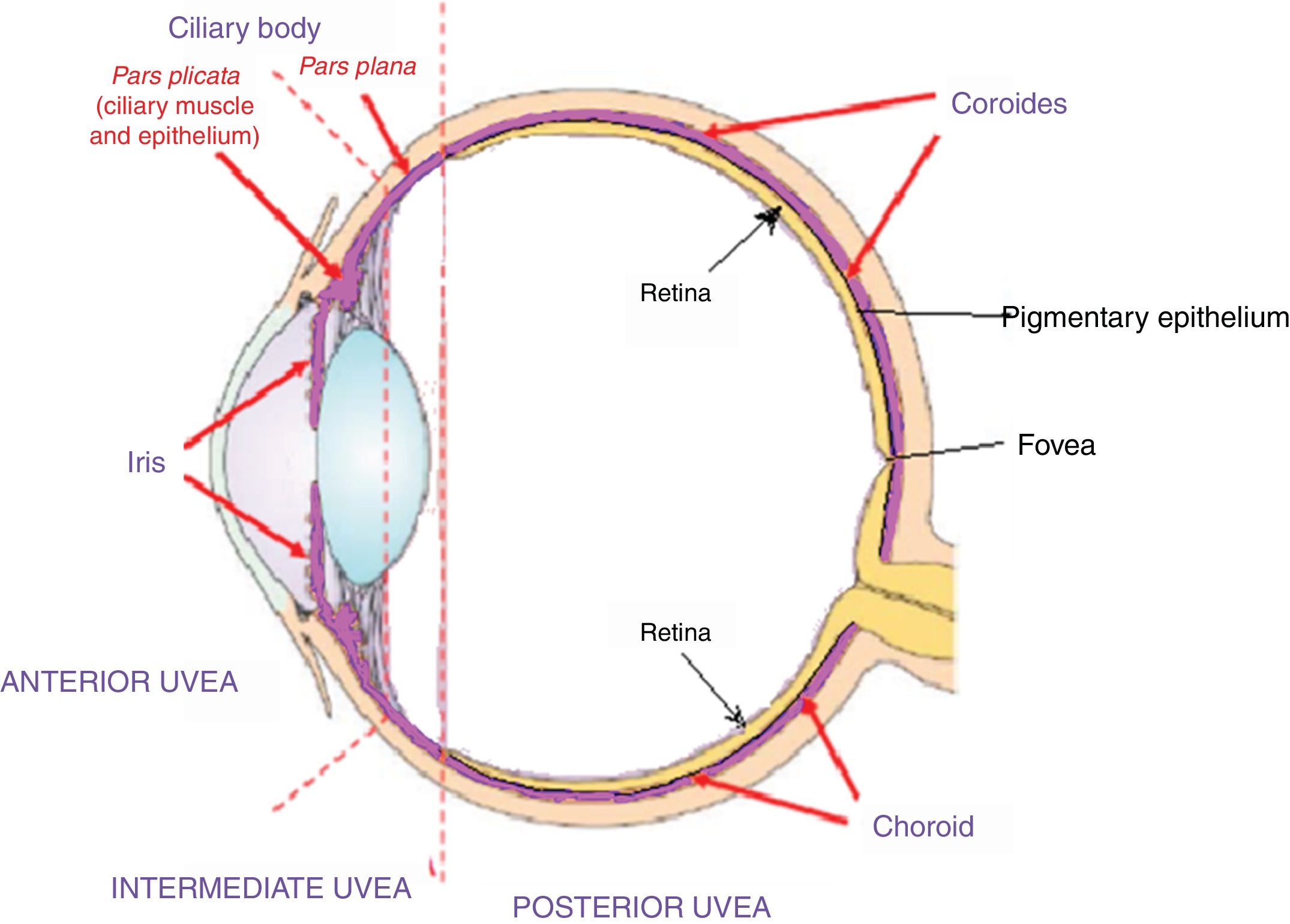
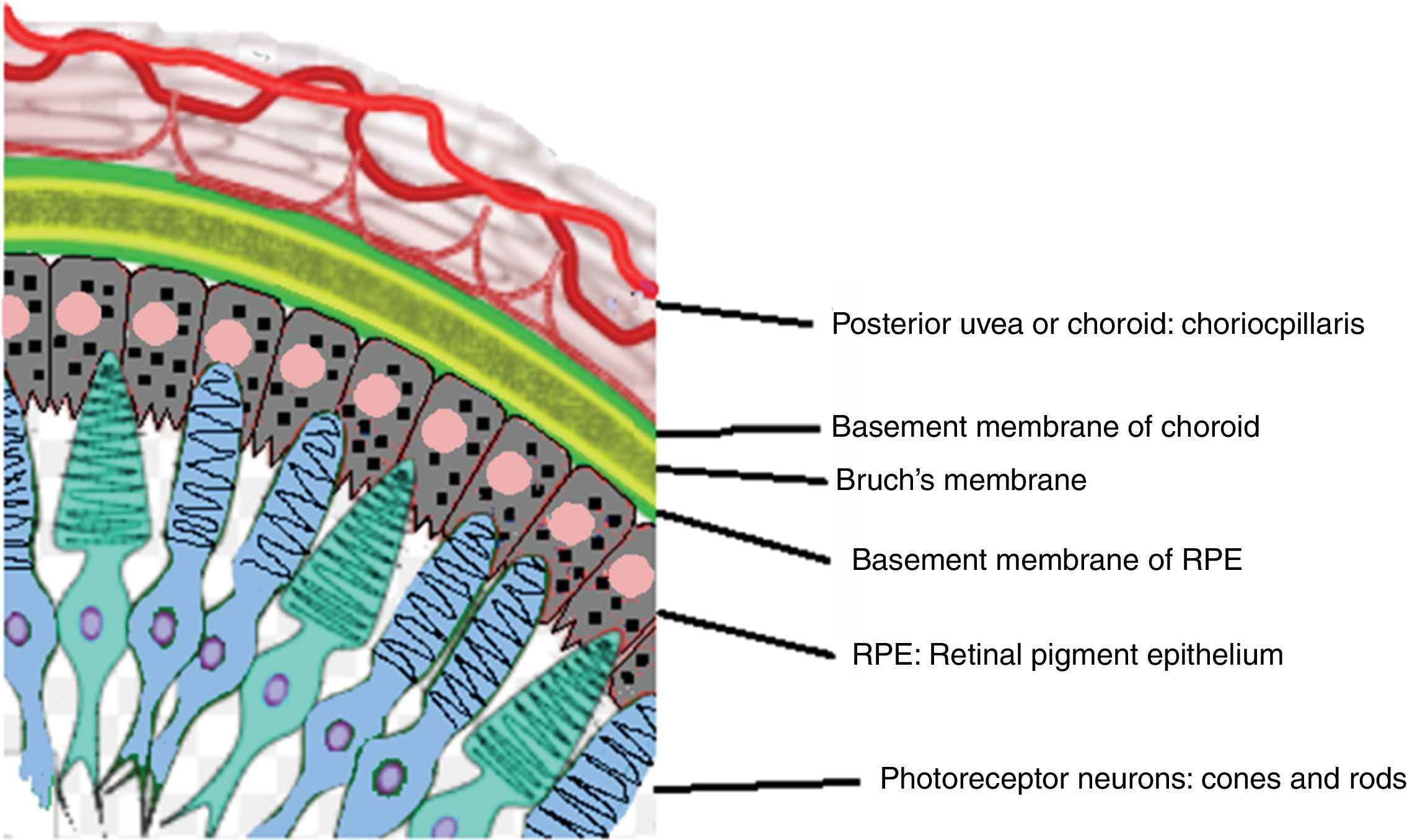
Acute uveitic phaseIt is considered the distinctive phase of VKH disease and can last for several weeks, in the course of which the majority of patients consult ophthalmology for presenting bilateral visual loss, usually asymmetric, due to diffuse choroiditis. In Fig. 5 is drawn the uveal tract in a cross-section of the eyeball, and the choroid or posterior uvea initially involved in VKH is indicated. Fig. 6 shows the drawing of a histological segment of the junction of the retinal pigment epithelium (RPE) and the choroid, which are the structures especially affected by autoimmune inflammation.
 which make up the anterior uvea, the pars plana or intermediate uvea and the choroid or posterior uvea. External limiting membrane.")
This cross-sectional drawing of the eye shows the uveal tract, highlighted in violet, with its 4 anatomical components: iris, ciliary body (pars plicata) which make up the anterior uvea, the pars plana or intermediate uvea and the choroid or posterior uvea. External limiting membrane.

Drawing that shows the anatomical relationship between the choriocapillaris framework of the choroid and the most external layer of the retina, the pigmentary epithelium. These structures are the target of the ocular inflammatory commitment in VKH disease.
In this acute uveitic phase the examiner can observe edema and hyperemia of the optic disc before observing cells in the vitreous and the classic bilateral exudative retinal detachment.5,6 The inflammatory involvement of the choroid alters the external hematoretinal barrier, formed by the Bruch’s membrane and the RPE, and causes accumulation of subretinal fluid and multiple retinal detachments, which can converge and form large bullous detachments. The presence of choroidal thickening of peripapillary predominance can be clearly observed by OCT,12–14 findings that correlate with inflammatory diffuse mononuclear cell infiltration. In this phase, the inflammation can affect the anterior chamber. When ciliary body edema occurs, the intraocular pressure can rise and cause acute glaucoma by closure of the angle between the base of the iris insertion and the cornea.2,6
Convalescent phaseThe convalescent phase occurs several weeks after the acute uveitic phase, it can last several months and is characterized by the appearance of choroidal and cutaneous depigmentation areas. Sugiura’s sign or perilimbal vitíligo is the earliest: it can appear in the first month of the disease.1,3 With the loss of RPE and choroidal melanocytes, the pale optic disc appears on a bright red-orange background, a color given by the depigmented choroid. This image evokes in the examiner the twilight, with its characteristic "brightness or glow of sunset", and constitutes the clinical sign known in the English literature as sunset glow fundus,3 which appears 2–3 months after the uveitic phase.1 In this phase, the ophthalmologist can also observe foci of hyperpigmentation due to migration of RPE cells, especially in Hispanic patients, and multiple small, rounded white-yellowish lesions that histologically correspond to Dalen-Fuchs nodules,6 which are aggregates of lymphocytes and pigment-laden macrophages that appear in the posterior pole between Bruch's membrane and the RPE.1
The depigmentation of the skin (vitiligo), eyelashes, eyebrows and hair (poliosis) and alopecia are mainly observed during this phase as a characteristic finding.3 These cutaneous manifestations are more common in Asian patients.5,15 The vitiligo is usually distributed symmetrically and affects the facial region, eyelids, trunk and sacral skin area.2
Chronic recurrent phaseIn the course of the convalescent phase, after months of treatment, some patients may evolve into the chronic phase, which is characterized by the appearance of recurrent granulomatous anterior uveitis, which leads to the development of nodules in the iris, focal atrophy of the iris and ocular hypotonia.1,4,7 During this phase, cases that have occurred with posterior choroidal inflammation, choroidal thickening and retinal detachments, demonstrated by indocyanin green angiography (ICGA)1 and OCT, have been reported.16 This chronic recurrent phase usually appears in the first 6 months of the disease, as a consequence of a rapid decrease or early suspension of corticotherapy.16,17
Complications derived from the chronic intraocular inflammation arise: the most frequent are cataracts and glaucoma, followed by subretinal neovascularization or fibrosis; less frequently, posterior synechiae, neovascularization of the optic disc, arteriovenous anastomoses and choroidal neovascularization occur.4,6,7 All these complications severely compromise the visual prognosis.
Etiology and pathogenesisThe pathophysiology of VKH has been widely studied in the last 2 decades; however, the precise etiology is not yet well established. It can be stated that the pathogenesis of VKH is multifactorial, and that its main target is the choroid layer of the eye. (Figs. 5 and 6). Although immunogenetic and environmental etiologies have been reported, the immune process that leads to VKH disease is an autoimmune response mediated by T cells against one or more antigenic components of the melanocytes in the eye, ear, skin, choroid plexus and brain.1,7,18 Several human leukocyte antigen (HLA) and non-HLA genes have been identified and related to VKH disease. Based on this differentiation, immunogenetic factors have been classified as associated and not associated with HLA.18,19
The non-HLA genes related to the disease are IL-23A, IL-23R, IL-17F, IL-27. Recent studies in experimental autoimmune uveitis, in a classical animal model, point to a central role of Th 17 cells in the pathogenesis of uveitis.20 These results in the animal model were confirmed in a study of patients with VKH, according to which it is demonstrated that the production of IL-17, the main cytokine of Th17 cells, is an essential part of the mechanism involved in the development of uveitis in patients with VKH. This study demonstrates that the reduced expression of IL-27, which is an anti-inflammatory cytokine that regulates the activation of Th 17 lymphocytes, results in increased expression of Th 17 in patients with VKH and active uveitis.20 The importance of these findings lies in the fact that the manipulation of IL-27 can offer new treatments for VKH. So far it is known that treatment with corticosteroids, by inducing a positive regulation of IL-27 and a negative regulation of IL-17, contribute to the resolution of intraocular inflammation.20
Several HLA genes that are related to VKH disease have also been identified, but the strength of association between these genes and the disease is not the same in different ethnic groups. Thus, for example, the association of VKH with HDL-DR4/DRw53 was found in Asian, North American natives and Hispanic patients.5,18 Conversely, the HLA-DRB1*0405 and HLA-DRB1*0410 alleles were strongly associated with VKH in Hindu patients.4
In addition to autoimmune responses against melanocytes, VKH disease appears to be caused by autoimmune activity against melanocyte-associated antigens. It has been demonstrated that antigens of the tyrosinase protein family and GP100, a protein expressed in the basement membrane of the melanosome are antigenic targets recognized by T cells of VKH patients with positive HLA DRB1*0405 and have been related to the cause and the VKH disease.5,18
KU-MEL-1 is another autoantigen that is widely expressed in most melanoma cell lines, samples of melanoma tissues and in cultured melanocytes.18 The antibody against KU-MEL-1 positive in serum showed a very significant association in VKH patients with positive HLA- DRB1*0405, compared with patients affected with other uveitis and with healthy controls.5,18 This association implies a primary function of KU-MEL-1 specific CD4 + T lymphocytes in the pathogenesis of VKH.
As for environmental and infectious factors, viral infections and autoimmune diseases have been correlated with VKH disease. Viruses such as Epstein-Barr and cytomegalovirus have been proposed as possible trigger factors of the disease.18 VKH has been reported after treatment with bacillus Calmette Guerin for melanoma, after surgery of metastatic melanoma and after traumatic skin wounds.2,6 Cases that associate VKH with hepatitis C therapy based on interferon alfa-2b combined or not with ribavirin are being reported.21
DiagnosisThe revised criteria for VKH have been widely accepted since their publication in 2001.3Table 2 shows the revised diagnostic criteria for VKH in the acute and chronic stages and accounts for its multisystemic nature. The diagnosis of VKH is mainly based on clinical findings and is complemented by images.
Diagnostic criteria for Vogt-Koyanagi-Harada disease.
| Complete VKH: criteria 1–5 must be present |
|---|
| There is no history of penetrating eye trauma or surgery that precede the initial onset of uveitis |
| No clinical or laboratory evidence suggesting other ocular disease entities. |
| Bilateral ocular involvement (depending on the stage of the disease at the time when the patient is examined): |
| 3a) Early manifestations |
| 1. Diffuse choroiditis (with or without anterior uveitis, vitreous inflammatory reaction, or hyperemia of the optic disc), characterized by: |
| a) Focal areas of subretinal fluid or |
| b) Bullous serous retinal detachments |
| 2. If clinical examination is doubtful, both a and b must be present: |
| a) Fluoroscein angiography: Focal areas of choroidal filling delay, multifocal punctiform leaks, accumulation of fluorescein in the subretinal fluid and staining of the optic nerve, and |
| b) Ultrasound: diffuse thickening of the choroid, with no evidence of posterior scleritis. |
| 3b) Late manifestation |
| 1) History suggestive of findings of 3a, with the following signs 2 and 3, or multiple signs 3: |
| 2) Ocular depigmentation: |
| a) Sunset glow fundus |
| b) Sugiura’s sign |
| 3) Other ocular signs: |
| a) Nummular chorioretinal depigmented scars or |
| b) Accumulation or migration of retinal pigment epithelium |
| c) Recurrent or chronic anterior uveítis |
| Neurological/auditory findings |
| 4a) Meningism (malaise, headache, nausea, abdominal pain, nuchal and back rigidity) |
| 4b) Tinnitus |
| 4c) Pleocytosis in cerebrospinal fluid |
| Dermatological manifestations (which do not precede the onset of neurological/ocular manifestations): |
| 5a) Alopecia |
| 5b) Poliosis |
| 5c) Vitiligo |
| Incomplete VKH: 1–3 and 4 or 5 criteria must be present |
| Probable VKH: isolated ocular disease, 1–3 criteria must be present |
There are 3 clinical entities: complete, incomplete and probable, derived from 5 criteria. As can be seen in Table 2, the absence of antecedents of penetrating trauma or eye surgery and of evidence indicative of other eye diseases is mandatory. In addition, bilateral ocular involvement indicating choroidal inflammation in the acute or chronic phase should be documented. The aforementioned findings comply with the diagnosis of probable VKH or isolated eye disease. The incomplete type requires neurological/auditory or dermal findings and the complete type requires both.5 In addition, the dermatological findings should not precede the commitment of the central nervous system or the eye disease.
The presentation of the disease in its complete, incomplete and probable forms shows varied prevalences derived from ethnic and geographical differences and from the duration of follow-up. For example, a prevalence of the complete type of 67 % is reported in China, while in the United States and Saudi Arabia the probable form is more common, in about 60 %.5
Complementary studiesIn most cases, when the patient has ocular and extraocular manifestations, the diagnosis of the disease is clinical.3 However, when the disease appears with visual compromise without extraocular involvement, several complementary tests have shown to be useful in confirming the diagnosis.1,22 Multimodal imaging has provided crucial information for the diagnosis, the evaluation of treatment and the supervision of retinochoroidal diseases, including VKH. Multimodal images encompass non-invasive methods: fundus photography, optical coherent tomography OCT, enhanced depth imaging OCT, ultrasound biomicroscopy (UBM), B-scan ultrasonography, fundus autofluorescence and invasive methods such as fundus fluorescein angiography (FFA) and ICGA.23 Their diagnostic role is clearly demonstrated in typical cases of VKH and is key in the exclusion of diseases that may enter into the differential diagnosis of atypical cases; in addition, multimodal images serve as a sign of prognostic value and have a fundamental role in monitoring intraocular inflammation, in the response to treatment and in the development of complications.
The findings of the FFA are very characteristic both in the acute phase and in the chronic phase of the disease. In the acute phase, during the initial stage of FFA, typically, multiple hyperfluorescent points appear in the RPE, which increase in size in the late stage. Such points represent accumulation of fluorescein in the spaces corresponding to retinal detachments. In the late stage the angiogram shows multiple hyperfluorescent multimodal images. In the majority of patients the FFA shows papillary hyperfluorescence and abnormal choroidal filling in patches.23 The FFA in the chronic phase of VKH is also very useful: it can show typical alterations in areas of atrophy or hyperplasia of RPE, choroidal neovascularization, retinochoroidal anastomosis and retinal or papillary neovascularization.2,23
ICGA is an appropriate method for studying choroidal diseases, including VKH disease, useful in early diagnosis, both in the acute phase and in the course of treatment and to assess the response obtained. The findings in the ICGA are useful to determine if there is active or persistent inflammation and, also, to define if there is resolution of the disease.1,7
OCT is useful for the diagnosis and monitoring of multimodal exudative macular images in the acute phase of the disease. The OCT facilitates the registration of images which are considered classic patterns of multimodal imaging in VKH.10,13
Through enhanced depth imaging OCT, it has been possible to measure the choroidal thickness at different stages of the disease and thus demonstrate a greater thickness in the acute phase and its decrease after treatment, so it could be useful as a marker of the degree of choroidal inflammation.12,17 In patients with long-term disease (longer than 6 months), it was observed a choroid significantly thinner than in the controls; in fact, this choroidal thinning, which is common in VKH, can only be measured accurately by enhanced depth imaging OCT: it is determined that the choroidal thickness is inversely proportional to the duration of the disease.17,24
UBM ultrasound and biomicroscopy provide images that are important for the diagnosis of the disease in its different phases, particularly in the acute stage.1,3 The UBM can show ciliochoroidal detachment with flattening of the anterior chamber in the initial phases of VKH.6
The MRI of the brain and orbits with contrast is useful when the patient consults early in the course of the prodromal or acute uveitic phases. The MRI of the brain allows to see meningeal enhancements and intraparenchymal focal lesions. The MRI of the orbits allows to differentiate the choroid and the sclera, and also identifies multimodal images. In cases of VKH it rules out primary scleral disease when diagnostic difficulties arise.2,25
The experts consider LP of non-routine limited practice for the diagnosis of VKH: it is useful in atypical cases or in patients who consult early with few ocular signs and positive meningeal signs.3,5,6
TreatmentThe treatment is based on the use of 3 elements: corticosteroids, immunosuppressive therapy (IST) and biological response modifiers.
CorticosteroidsIn VKH the treatment is aimed at suppressing the acute choroidal inflammation with the early onset of systemic corticosteroids at high and sustained doses for 4–6 months, followed by a progressive decrease.3
Recently, it has been described a reduction in the choroidal and retinal thickness after the initiation of intravenous methylprednisolone for 3–5 days, at doses ranging between 200 mg/day and one gram/day, followed by oral prednisolone 1 mg/kg per day.5,26 This regimen of corticosteroids consisting in high and early doses allows a resolution of the disease with fewer complications than with later and lower doses.26 Even so, the chronic stage of the disease can develop in up to a third of patients.
Local steroid injections have also been used and they include their transeptal application, intravitreal injections and sustained-release corticosteroid implants whose efficacy and safety have not been clearly defined.27,28 For the inflammation of the anterior chamber, topical 1 % prednisolone and cycloplegic agents are indicated, either in the chronic or initial recurrent phase, in order to reduce pain and inflammation and to prevent synechiae. Topical corticosteroids should be discontinued as the inflammation decreases.2,5
Early use of high doses of systemic corticosteroids has also been shown to be effective for the treatment of auditory manifestations of VKH. Of the cases of VKH with hearing problems, 75 % returned to normality; however, although very rarely, VKH disease can cause profound hearing loss.11
Immunosuppressive therapyThe usefulness of IST has been widely demonstrated in patients with autoimmune systemic problems and its use allows to reduce the side effects of prolonged administration of corticosteroids. The American Uveitis Society and the consensus panel of the International Uveitis Study Group accepted the need for IST in the VKH because there is a significantly better functional outcome with its early initiation.2 In a retrospective cohort study conducted in Chile, the authors found significantly better visual evolution with the early initiation of IST in a subgroup of patients with VKH with severe compromise of visual acuity.29
Cyclosporine A, a calcineurin inhibitor that more specifically targets T cells, is an appropriate choice in the treatment of VKH. Cyclosporine has been the most widely used immunosuppressive agent in the care of patients with this disease.30 With an action similar to that of cyclosporine, tacrolimus, a T-cell inhibitor, is another potential cytostatic therapeutic option. Among the antimetabolite agents, mofetil mycophenolate and azathioprine, T cells and B cells suppressors, are reliable treatment options in VKH patients.31 Methotrexate has demonstrate a good safety profile in the long term, with clinical efficacy in children and adults.30 The synergistic effects of the triple agent IST with the combination of oral prednisolone, azathioprine and cyclosporine for the rapid control of inflammation and a good visual outcome in severe and refractory cases of VKH have also been reported.32
In summary, IST therapy combined with systemic corticosteroids significantly reduced the recurrences of uveitis, the development of late complications and improved the visual outcome compared with another group of VKH patients treated with corticosteroid monotherapy or with late addition of IST.
Biological response modifiersIn addition to corticosteroids and IST, new biological response modifier therapies have been opening their way in the treatment of uveitis. Monoclonal antibodies that block specific inflammatory cytokines, such as infliximab32 and adalimumab33 which are directed against the tumor necrosis factor α (TNF-alpha) and the anti-CD20 rituximab,34 are being used in uveitis refractory to immunomodulatory therapy.
The vascular endothelial growth factor (VEGF), a cytokine that promotes endothelial cell proliferation and increases vascular permeability, plays an important role in the pathogenesis of uveitic complications, such as cystoid macular edema, choroidal neovascularization and neovascularization of the retina.35 It has been demonstrated an overexpression of the levels of VEGF in serum and aqueous humor of patients with different types of uveitis, including VKH. Intravitreal application of anti-VEGF antibodies, such as bevacizumab and ranibizumab, inhibits choroidal neovascularization and reduces the serous retinal detachments in VKH patients, therefore, they are now considered as adjuvant therapy to conventional systemic therapy.18
DiscussionVKH is a disease that has been well studied, with precise diagnostic criteria. Despite having a regular and consistent clinical presentation pattern, it is little known in Colombia. During the search period, case reports or reviews of VKH were not found in the available Colombian medical literature. This may indicate that the disease occurs and is underdiagnosed or unreported.
The clinical diagnosis of VKH can offer difficulties for several reasons. First, the disease has 4 chronological phases and at the time of the initial consultation the patients may be in any phase of the disease and the demonstration of choroidomeningeal inflammation and its characterization in the corresponding phase becomes a real challenge. Second, when the acute uveitic phase occurs, the patients alarmed by the visual alterations consult for the first time, usually ophthalmology. However, it is important to keep in mind that this phase of acute ocular inflammation is frequently established when the prodromal phase is still on course, thus forming a clinical picture characterized by headache, dysacusia, tinnitus, meningism and ocular alterations. This presentation of the disease constitutes a diagnostic challenge for the medical group. Its clinical approach should include the study of the ophthalmological alteration and the neurological and otovestibular involvement, simultaneously. It was in these conditions of clinical concomitance between the prodromal phase and the acute uveitic phase when the patient of this case consulted. Therefore, in cases like this, it is considered important to recognize the various presentations of VKH emphasizing the clinical and paraclinical characteristics of the 2 initial phases in order to diagnose the disease early.
Regarding the diagnostic criteria, ophthalmologists of endemic areas of VKH and experts in uveitis of specialized reference centers question the need to practice LP as a diagnostic procedure, considering that the diagnosis of the disease is fundamentally clinical and, in doubtful cases, the AGF and OCT images of the macula and optic nerve help to confirm the clinical impression and, therefore, it is not advisable to expose patients to the side effects of LP.
Against this opinion, the authors of the present work consider that, on the one hand, the diagnosis of choroiditis during the acute uveitic phase of VKH disease is not as evident in a first ophthalmological examination and, on the other hand, although the classic acute choroiditis of VKH, with or without retinal detachments, is observed in the ophthalmological examination, if the patient has headache and signs of meningeal irritation the neurological approach is necessary and the LP is mandatory. In fact, the uveomeningoencephalic syndrome poses a very broad etiological spectrum that includes several infectious, neoplastic, paraneoplastic and autoimmune diseases. Consequently, the study of the CSF is key to give precision and diagnostic certainty. If lymphocytic pleocytosis in CSF is demonstrated, in consonance with aseptic meningitis, a criterion that allows to initiate immunosuppressive treatment is met. Even, a new LP should be performed to a patient with established diagnosis of VKH, immunosuppressed with treatment, who in the course of the evolution presents headache and meningism, in order to rule out an opportunistic neuroinfection or recurrent aseptic meningitis. In a risk-benefit balance, the benefit obtained from LP is much greater compared to its possible complications; the fear of side effects derived from an LP does not appear to be a valid reason to hinder its realization.
ConclusionsThe topic review provided relevant updated information to answer the question formulated by the research group and draw the following conclusions:
- 1
There are clear and well-defined diagnostic criteria for VKH in its different phases that allow us to provide sufficient knowledge to make the clinical diagnosis of VKH in every middle-aged patient who consults for myodesopsia, blurred vision, dyschromatopsia, headache, meningism and auditory-vestibular symptoms. This is a first form of valid diagnostic intervention according to experts; nevertheless, the research group of the present work considers that the clinical diagnosis is admissible in endemic areas where the disease is frequent, but not in areas where the disease is occasional and little known.
- 2
When the clinical diagnosis of VKH disease becomes difficult in cases of atypical presentations or when the case occurs in non-endemic areas, focusing the study of the case starting from a syndromic diagnosis is a valid and appropriate form of intervention. This is how the diagnosis of uveomeningoencephalic syndrome alerts the medical care team, implies hospitalization in a center of high complexity, facilitates the simultaneous attention of a multidisciplinary team and allows to speed up the paraclinical tests. The second form of intervention was given to the present case where the results of FFA, OCT, fundus photograph, MRI and LP allowed to establish the diagnosis of VKH disease. For the research group, this second form of intervention is relevant and useful in regions such as ours, where the disease is rare and little known. In conclusion, in Colombia it is necessary to use paraclinical tests to make the diagnosis of VKH.
- 3
The updated review of the etiology and physiopathogenesis of VKH disease demonstrates its autoimmune and multisystemic nature, which requires an interspecialized medical approach. The participation of ophthalmology, internal medicine, neurology, rheumatology, otorhinolaryngology and dermatology becomes relevant and necessary for diagnostic and therapeutic purposes.
- 4
As for treatment, the vigorous use of corticosteroids in the acute uveitic phase is recommended. Evidence-based therapy recommends the use of methylprednisolone pulses of 1 g intravenously per day for 3–5 days, followed by oral prednisolone 1 mg/kg per day, always adding IST early, with cyclosporine as the most widely used drug. The use of methotrexate, tacrolimus, azathioprine or mycophenolate mofetil is also recommended. Low doses of corticosteroids or their rapid withdrawal are associated with severe ocular complications. Intravitreal application of anti-VEGF antibodies, such as bevacizumab and ranibizumab is recommended because it has been seen a reduction in serous retinal detachments in patients with VKH, inhibits choroidal neovascularization and improves visual prognosis. The adjustment of treatment with corticosteroid and IST requires strict control by ophthalmology, rheumatology or internal medicine, and it should not be suspended before one year, according with the clinical and imaging evolution.
The authors declare they do not have any conflict of interest.
Please cite this article as: Betancourt R, Betancourt SA, Soler G, Mantilla RD, Castillo GA. Enfermedad de Vogt Koyanagi Harada. Reporte de un caso y revisión de la literatura. Rev Colomb Reumatol. 2020;27:50–60.
