


Las enfermedades metabólicas hereditarias son enfermedades mendelianas que agrupan a un colectivo de más de 600 patologías clasificadas en función de la vía metabólica alterada y su patogénesis. La mayoría se diagnostican posnatalmente tras el reconocimiento por parte del clínico de una serie de síntomas sugestivos de enfermedad. Los laboratorios de genética bioquímica y molecular están implicados en el reconocimiento de estas enfermedades ya que el análisis de metabolitos, proteínas y genes son claves para su diagnóstico. En este artículo se revisa cómo (abordaje) se diagnostican estas enfermedades y las técnicas bioquímicas y genéticas de laboratorio comúnmente utilizadas. Es en el ámbito de la identificación de metabolitos (espectrometría de masas) y detección de mutaciones (secuenciación masiva) donde el impacto de las nuevas tecnologías en estos últimos años ha sido espectacular, lo que ha facilitado el diagnóstico rápido y con pruebas poco invasivas y facilitará en el futuro el cribado poblacional.
Inherited metabolic diseases are a group of more than 600 diseases classified according to the altered metabolic pathway and their pathogenesis. Most are diagnosed postnatally after recognition by a number of clinical symptoms suggestive of disease. Laboratories of biochemical and molecular genetics are involved in the recognition of these diseases as the analysis of metabolites, proteins and genes are key for diagnosis. This article reviews how (approach) these diseases are diagnosed and the biochemical and genetic laboratory techniques commonly used. It is in the area of identification of metabolites (mass spectrometry) and mutation detection (massive sequencing) where the impact of new technology in recent years has been spectacular, which facilitated rapid diagnosis with minimally invasive tests and will facilitate future population screening.
Las enfermedades metabólicas hereditarias (EMH) son enfermedades mendelianas (www.ncbi.nlm.nih.gov/Omim/mimstats.html), que agrupan a un colectivo de más de 600 patologías diferentes clasificadas en función de la vía metabólica alterada y su patogénesis. La mayor parte cumplen el criterio de enfermedad rara según la definición de la Unión Europea: enfermedades raras, minoritarias, huérfanas o enfermedades poco frecuentes, incluidas las de origen genético, son aquellas enfermedades con peligro de muerte o de invalidez crónica que tienen una prevalencia menor de 5 casos por cada 10.000 habitantes. Aunque individualmente son poco frecuentes, colectivamente son numerosas, contribuyendo de una forma muy significativa al número total de enfermos en la sociedad.
Las EMH se definen como trastornos genéticamente determinados en la síntesis y/o función de moléculas proteicas. Gran parte de estas proteínas son enzimas cuyo defecto da lugar a un bloqueo en la vía metabólica implicada y la consiguiente acumulación en fluidos corporales del sustrato de la reacción y otros metabolitos derivados. Este aumento no fisiológico de estos compuestos, en muchos casos tóxicos, así como la disminución del producto de la reacción, están implicados en los mecanismos fisiopatológicos de la enfermedad1.
La mayoría se heredan de forma autosómica recesiva, alrededor del 20% tienen herencia autosómica dominante, el 10% una herencia ligada al cromosoma X y una parte importante de las denominadas enfermedades mitocondriales tienen una herencia materna ya que afectan al ADN mitocondrial.
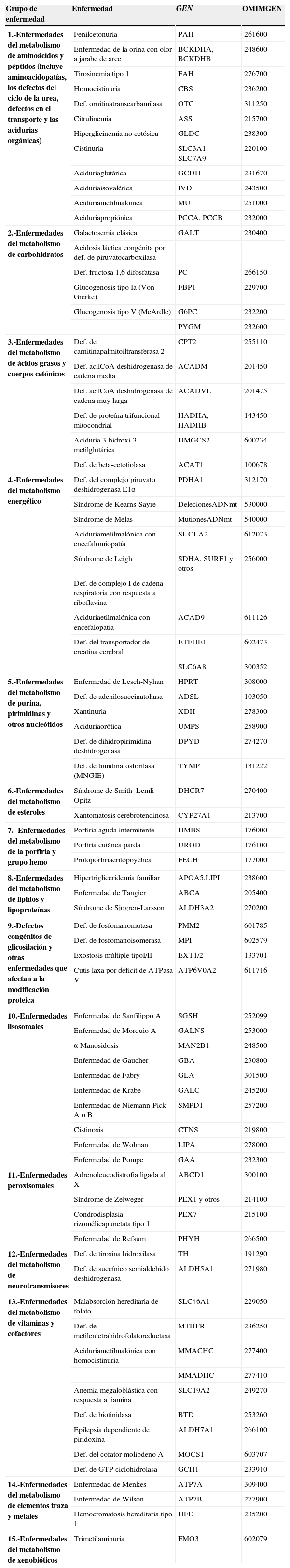
Recientemente, la Society for the Study of Inborn Errors of Metabolism ha elaborado una clasificación de los EMH, agrupando las enfermedades en 15 grandes grupos, según afecten a moléculas sencillas del metabolismo intermediario o energético o a moléculas complejas localizadas en diferentes organelas celulares (tabla 1)2.
Clasificación de las EMHs según la SSIEM y enumeración de las más frecuentes o conocidas en cada grupo
| Grupo de enfermedad | Enfermedad | GEN | OMIMGEN |
|---|---|---|---|
| 1.-Enfermedades del metabolismo de aminoácidos y péptidos (incluye aminoacidopatías, los defectos del ciclo de la urea, defectos en el transporte y las acidurias orgánicas) | Fenilcetonuria | PAH | 261600 |
| Enfermedad de la orina con olor a jarabe de arce | BCKDHA, BCKDHB | 248600 | |
| Tirosinemia tipo 1 | FAH | 276700 | |
| Homocistinuria | CBS | 236200 | |
| Def. ornitinatranscarbamilasa | OTC | 311250 | |
| Citrulinemia | ASS | 215700 | |
| Hiperglicinemia no cetósica | GLDC | 238300 | |
| Cistinuria | SLC3A1, SLC7A9 | 220100 | |
| Aciduriaglutárica | GCDH | 231670 | |
| Aciduriaisovalérica | IVD | 243500 | |
| Aciduriametilmalónica | MUT | 251000 | |
| Aciduriapropiónica | PCCA, PCCB | 232000 | |
| 2.-Enfermedades del metabolismo de carbohidratos | Galactosemia clásica | GALT | 230400 |
| Acidosis láctica congénita por def. de piruvatocarboxilasa | |||
| Def. fructosa 1,6 difosfatasa | PC | 266150 | |
| Glucogenosis tipo Ia (Von Gierke) | FBP1 | 229700 | |
| Glucogenosis tipo V (McArdle) | G6PC | 232200 | |
| PYGM | 232600 | ||
| 3.-Enfermedades del metabolismo de ácidos grasos y cuerpos cetónicos | Def. de carnitinapalmitoiltransferasa 2 | CPT2 | 255110 |
| Def. acilCoA deshidrogenasa de cadena media | ACADM | 201450 | |
| Def. acilCoA deshidrogenasa de cadena muy larga | ACADVL | 201475 | |
| Def. de proteína trifuncional mitocondrial | HADHA, HADHB | 143450 | |
| Aciduria 3-hidroxi-3-metilglutárica | HMGCS2 | 600234 | |
| Def. de beta-cetotiolasa | ACAT1 | 100678 | |
| 4.-Enfermedades del metabolismo energético | Def. del complejo piruvato deshidrogenasa E1α | PDHA1 | 312170 |
| Síndrome de Kearns-Sayre | DelecionesADNmt | 530000 | |
| Síndrome de Melas | MutionesADNmt | 540000 | |
| Aciduriametilmalónica con encefalomiopatía | SUCLA2 | 612073 | |
| Síndrome de Leigh | SDHA, SURF1 y otros | 256000 | |
| Def. de complejo I de cadena respiratoria con respuesta a riboflavina | |||
| Aciduriaetilmalónica con encefalopatía | ACAD9 | 611126 | |
| Def. del transportador de creatina cerebral | ETFHE1 | 602473 | |
| SLC6A8 | 300352 | ||
| 5.-Enfermedades del metabolismo de purina, pirimidinas y otros nucleótidos | Enfermedad de Lesch-Nyhan | HPRT | 308000 |
| Def. de adenilosuccinatoliasa | ADSL | 103050 | |
| Xantinuria | XDH | 278300 | |
| Aciduriaorótica | UMPS | 258900 | |
| Def. de dihidropirimidina deshidrogenasa | DPYD | 274270 | |
| Def. de timidinafosforilasa (MNGIE) | TYMP | 131222 | |
| 6.-Enfermedades del metabolismo de esteroles | Síndrome de Smith–Lemli-Opitz | DHCR7 | 270400 |
| Xantomatosis cerebrotendinosa | CYP27A1 | 213700 | |
| 7.- Enfermedades del metabolismo de la porfiria y grupo hemo | Porfiria aguda intermitente | HMBS | 176000 |
| Porfiria cutánea parda | UROD | 176100 | |
| Protoporfiriaeritopoyética | FECH | 177000 | |
| 8.-Enfermedades del metabolismo de lípidos y lipoproteínas | Hipertrigliceridemia familiar | APOA5,LIPI | 238600 |
| Enfermedad de Tangier | ABCA | 205400 | |
| Síndrome de Sjogren-Larsson | ALDH3A2 | 270200 | |
| 9.-Defectos congénitos de glicosilación y otras enfermedades que afectan a la modificación proteica | Def. de fosfomanomutasa | PMM2 | 601785 |
| Def. de fosfomanoisomerasa | MPI | 602579 | |
| Exostosis múltiple tipoI/II | EXT1/2 | 133701 | |
| Cutis laxa por déficit de ATPasa V | ATP6V0A2 | 611716 | |
| 10.-Enfermedades lisosomales | Enfermedad de Sanfilippo A | SGSH | 252099 |
| Enfermedad de Morquio A | GALNS | 253000 | |
| α-Manosidosis | MAN2B1 | 248500 | |
| Enfermedad de Gaucher | GBA | 230800 | |
| Enfermedad de Fabry | GLA | 301500 | |
| Enfermedad de Krabe | GALC | 245200 | |
| Enfermedad de Niemann-Pick A o B | SMPD1 | 257200 | |
| Cistinosis | CTNS | 219800 | |
| Enfermedad de Wolman | LIPA | 278000 | |
| Enfermedad de Pompe | GAA | 232300 | |
| 11.-Enfermedades peroxisomales | Adrenoleucodistrofia ligada al X | ABCD1 | 300100 |
| Síndrome de Zelweger | PEX1 y otros | 214100 | |
| Condrodisplasia rizomélicapunctata tipo 1 | PEX7 | 215100 | |
| Enfermedad de Refsum | PHYH | 266500 | |
| 12.-Enfermedades del metabolismo de neurotransmisores | Def. de tirosina hidroxilasa | TH | 191290 |
| Def. de succínico semialdehido deshidrogenasa | ALDH5A1 | 271980 | |
| 13.-Enfermedades del metabolismo de vitaminas y cofactores | Malabsorción hereditaria de folato | SLC46A1 | 229050 |
| Def. de metilentetrahidrofolatoreductasa | MTHFR | 236250 | |
| Aciduriametilmalónica con homocistinuria | MMACHC | 277400 | |
| MMADHC | 277410 | ||
| Anemia megaloblástica con respuesta a tiamina | SLC19A2 | 249270 | |
| Def. de biotinidasa | BTD | 253260 | |
| Epilepsia dependiente de piridoxina | ALDH7A1 | 266100 | |
| Def. del cofator molibdeno A | MOCS1 | 603707 | |
| Def. de GTP ciclohidrolasa | GCH1 | 233910 | |
| 14.-Enfermedades del metabolismo de elementos traza y metales | Enfermedad de Menkes | ATP7A | 309400 |
| Enfermedad de Wilson | ATP7B | 277900 | |
| Hemocromatosis hereditaria tipo 1 | HFE | 235200 | |
| 15.-Enfermedades del metabolismo de xenobióticos | Trimetilaminuria | FMO3 | 602079 |
La mayoría de las EMH se diagnostican posnatalmente tras el reconocimiento por parte del clínico de una serie de síntomas específicos y sugestivos de enfermedad metabólica. El comienzo puede ser a cualquier edad, aunque una gran parte de los pacientes presentan la enfermedad antes del año de vida. Suele comprometer a más de un sistema simultáneamente siendo el sistema nervioso central el más comúnmente afectado. El diagnóstico exacto de la proteína/gen afectado, permite el acceso a opciones preventivas para la familia que deben darse a conocer a través del asesoramiento genético. Previo consentimiento informado al paciente o a los padres o tutores del paciente u otros miembros de la familia, se buscarían posibles miembros afectos todavía asintomáticos, sobre todo si pueden beneficiarse de un tratamiento precoz, se podría hacer el diagnóstico de portadores de la enfermedad entre familiares, y se aportaría información sobre el tipo de herencia, riesgo de recurrencia, pronóstico del curso clínico de la enfermedad (si se conoce) y opciones reproductivas (diagnóstico prenatal y preimplantacional). Existe una pequeña parte de las EMH (10%) y algunas endocrinopatías que pueden detectarse presintomáticamente en los programas de cribado neonatal. El objetivo es identificar pacientes de pocos días de vida mediante pruebas analíticas poco costosas para intervenir precozmente y evitar, en lo posible, los efectos de la enfermedad.
Es en el ámbito de la identificación de metabolitos y detección de mutaciones donde el impacto de las nuevas tecnologías en estos últimos años ha sido espectacular, lo que ha facilitado el diagnóstico rápido y cada vez menos invasivo.
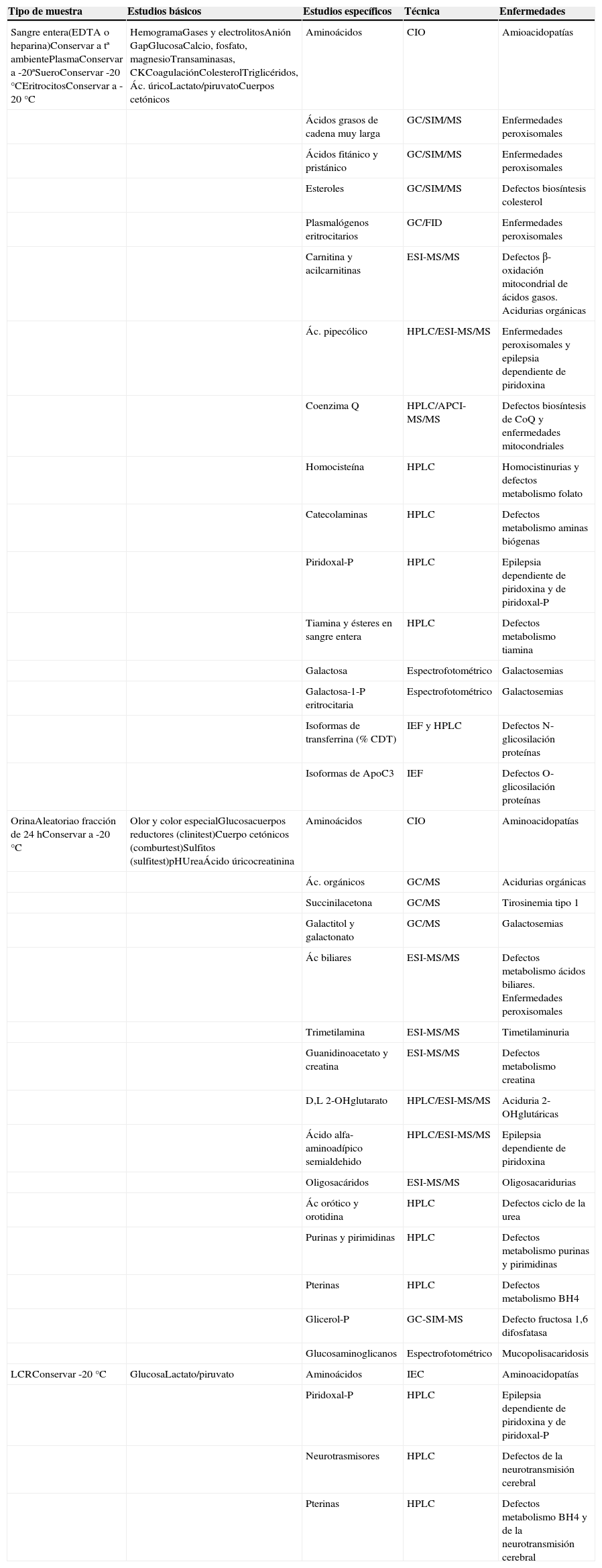
El laboratorio en el diagnóstico de las EMHLa mayoría de las EMH se diagnostican posnatalmente a partir de una sospecha diagnóstica realizada por el médico que reconoce una serie de síntomas y signos compatibles. Es conveniente un cribado clínico revisando los algoritmos diagnósticos de EMH en niños y adultos antes de iniciar investigaciones bioquímicas complicadas3. Es importante saber qué tipo de muestra es necesaria según la sospecha: sangre entera con anticoagulante, plasma o suero, orina de una micción o de 24 horas y líquido cefalorraquídeo (LCR). Cuándo debe tomarse, de forma urgente ante el cuadro agudo y antes de iniciar tratamientos que interfieran y cómo debe conservarse (tabla 2). La sospecha clínica inicial de un EMH exige la realización de una serie de estudios básicos que pueden hacerse en el hospital (tabla 2). Con estos resultados se establece una estrategia diagnóstica en coordinación con el laboratorio de genética bioquímica, que realizará otros análisis más específicos (tabla 2), a ser posible en las mismas muestras en las que se realizaron las pruebas básicas.
Tipo de muestra, estudios bioquímicos básicos y específicos (metabolitos) y técnica utilizada en el diagnóstico de EMH grupo
| Tipo de muestra | Estudios básicos | Estudios específicos | Técnica | Enfermedades |
|---|---|---|---|---|
| Sangre entera(EDTA o heparina)Conservar a tª ambientePlasmaConservar a -20ªSueroConservar -20°CEritrocitosConservar a -20°C | HemogramaGases y electrolitosAnión GapGlucosaCalcio, fosfato, magnesioTransaminasas, CKCoagulaciónColesterolTriglicéridos, Ác. úricoLactato/piruvatoCuerpos cetónicos | Aminoácidos | CIO | Amioacidopatías |
| Ácidos grasos de cadena muy larga | GC/SIM/MS | Enfermedades peroxisomales | ||
| Ácidos fitánico y pristánico | GC/SIM/MS | Enfermedades peroxisomales | ||
| Esteroles | GC/SIM/MS | Defectos biosíntesis colesterol | ||
| Plasmalógenos eritrocitarios | GC/FID | Enfermedades peroxisomales | ||
| Carnitina y acilcarnitinas | ESI-MS/MS | Defectos β-oxidación mitocondrial de ácidos gasos. Acidurias orgánicas | ||
| Ác. pipecólico | HPLC/ESI-MS/MS | Enfermedades peroxisomales y epilepsia dependiente de piridoxina | ||
| Coenzima Q | HPLC/APCI-MS/MS | Defectos biosíntesis de CoQ y enfermedades mitocondriales | ||
| Homocisteína | HPLC | Homocistinurias y defectos metabolismo folato | ||
| Catecolaminas | HPLC | Defectos metabolismo aminas biógenas | ||
| Piridoxal-P | HPLC | Epilepsia dependiente de piridoxina y de piridoxal-P | ||
| Tiamina y ésteres en sangre entera | HPLC | Defectos metabolismo tiamina | ||
| Galactosa | Espectrofotométrico | Galactosemias | ||
| Galactosa-1-P eritrocitaria | Espectrofotométrico | Galactosemias | ||
| Isoformas de transferrina (% CDT) | IEF y HPLC | Defectos N-glicosilación proteínas | ||
| Isoformas de ApoC3 | IEF | Defectos O-glicosilación proteínas | ||
| OrinaAleatoriao fracción de 24hConservar a -20°C | Olor y color especialGlucosacuerpos reductores (clinitest)Cuerpo cetónicos (comburtest)Sulfitos (sulfitest)pHUreaÁcido úricocreatinina | Aminoácidos | CIO | Aminoacidopatías |
| Ác. orgánicos | GC/MS | Acidurias orgánicas | ||
| Succinilacetona | GC/MS | Tirosinemia tipo 1 | ||
| Galactitol y galactonato | GC/MS | Galactosemias | ||
| Ác biliares | ESI-MS/MS | Defectos metabolismo ácidos biliares. Enfermedades peroxisomales | ||
| Trimetilamina | ESI-MS/MS | Timetilaminuria | ||
| Guanidinoacetato y creatina | ESI-MS/MS | Defectos metabolismo creatina | ||
| D,L 2-OHglutarato | HPLC/ESI-MS/MS | Aciduria 2-OHglutáricas | ||
| Ácido alfa-aminoadípico semialdehido | HPLC/ESI-MS/MS | Epilepsia dependiente de piridoxina | ||
| Oligosacáridos | ESI-MS/MS | Oligosacaridurias | ||
| Ác orótico y orotidina | HPLC | Defectos ciclo de la urea | ||
| Purinas y pirimidinas | HPLC | Defectos metabolismo purinas y pirimidinas | ||
| Pterinas | HPLC | Defectos metabolismo BH4 | ||
| Glicerol-P | GC-SIM-MS | Defecto fructosa 1,6 difosfatasa | ||
| Glucosaminoglicanos | Espectrofotométrico | Mucopolisacaridosis | ||
| LCRConservar -20°C | GlucosaLactato/piruvato | Aminoácidos | IEC | Aminoacidopatías |
| Piridoxal-P | HPLC | Epilepsia dependiente de piridoxina y de piridoxal-P | ||
| Neurotrasmisores | HPLC | Defectos de la neurotransmisión cerebral | ||
| Pterinas | HPLC | Defectos metabolismo BH4 y de la neurotransmisión cerebral |
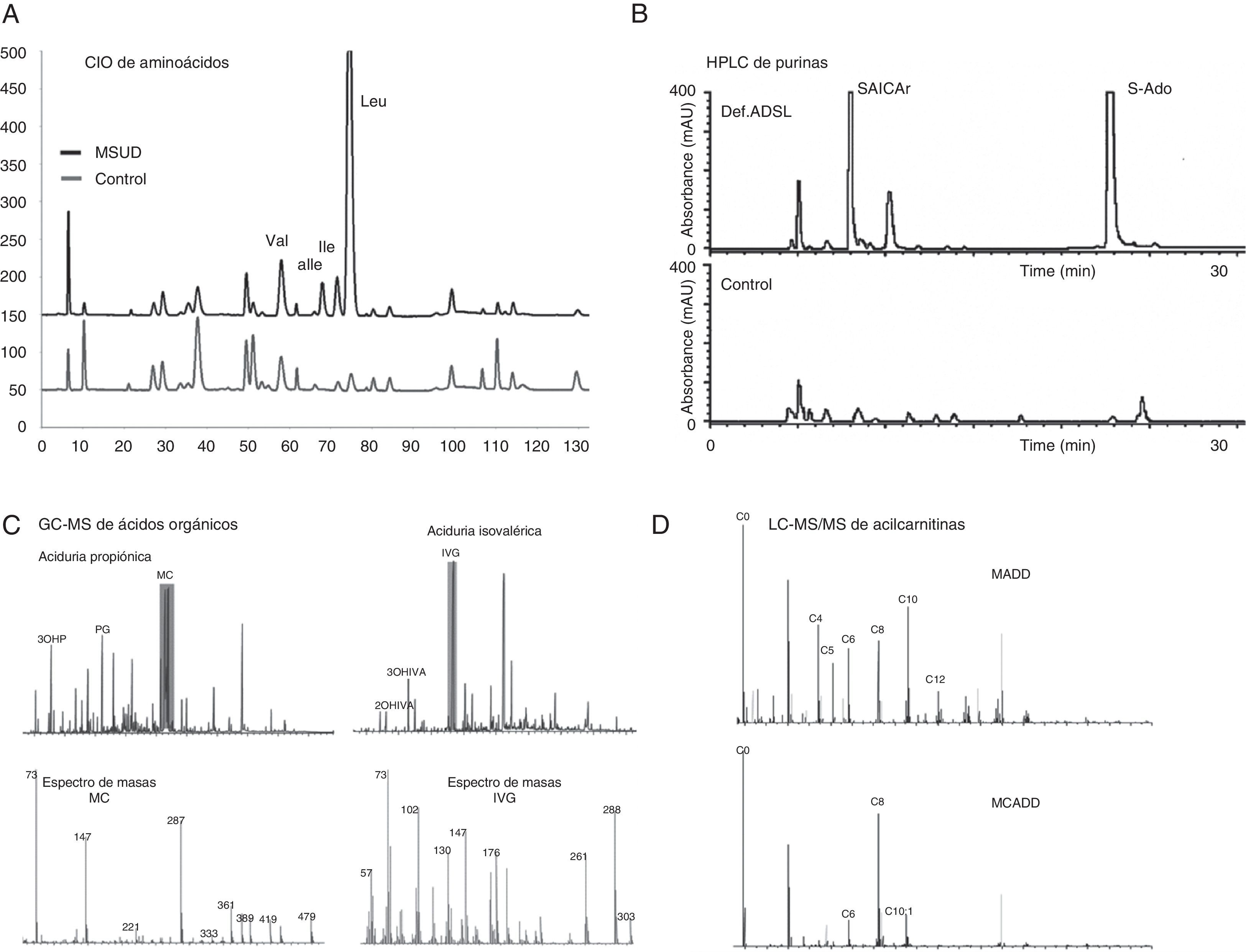
Estudio de las vías metabólicas (metabolitos). La cuantificación de metabolitos en fluidos fisiológicos e identificación de un perfil metabólico concreto es, en muchos casos, diagnóstico de una EMH y en otros permite orientar una sospecha. Se utilizan diversas técnicas analíticas, fundamentalmente cromatografía y espectrometría de masas, cada una de ellas es específica para un grupo de metabolitos. Para valorar los resultados, es importante tener bien establecidos los intervalos control según edad y conocer edad, alimentación y medicación en el momento de la toma de la muestra al paciente. La cromatografía de intercambio iónico (CIO) es el método de elección para la separación y cuantificación de aminoácidos4. Está basado en la separación de los aminoácidos en función de su carga eléctrica en una columna de intercambio catiónico, elución de los mismos secuencialmente utilizando tampones de litio a diferentes pH y detección por colorimetría tiñendo con ninhidrina. Es una técnica muy reproducible y sensible. Todas las aminoacidopatías pueden diagnosticarse midiendo aminoácidos en fluidos biológicos (fig. 1A). El análisis de aminoácidos también se realiza para monitorización del tratamiento de numerosas EMH y se utiliza como marcador del estatus nutricional y de la funcionalidad de órganos como hígado, riñón, intestino etc… La cromatografía líquida de alta resolución (high performance liquid chromatography [HPLC]) es un método muy versátil, también basado en la separación de metabolitos según sus propiedades físico-químicas. Se hace pasar las moléculas a través de una columna cromatográfica (fase estacionaria) bombeadas por un líquido o solvente (fase móvil) aplicando presión alta. El metabolito se identifica según su tiempo de retención y se detecta y cuantifica con distintos detectores. La detección electroquímica se utiliza en el análisis de neurotransmisores en LCR5, para el diagnóstico de, p.ej., las distonías con respuesta a dopa por defectos de tirosina hidroxilasa (síndrome de Segawa) o GTP ciclohidrolasa, que cursan con disminución de ácido homovanílico y/o ácido hidroxiindolacético. Con el detector de fluorescencia se mide la homocisteína6 y las pterinas7, estos últimos metabolitos alterados en las hiperfenilalaninemias por defectos en la síntesis y regeneración del cofactor BH4 de la fenilalanina hidroxilasa. Para la cuantificación de purinas y pirimidinas se utiliza el detector diodearray (UV/visible)8 (fig. 1B).
 Cromatograma de aminoácidos en plasma por CIO de un paciente de jarabe de arce (MSUD), se observa el aumento de los aminoácidos marcadores de la enfermedad valina (Val), aloisoleucina (aIle), isoleucina (Ile) y leucina (Leu). B) Cromatograma de purinas en líquido cefaloraquídeo (LCR) por HPLC de un paciente con deficiencia en adenilosuccinato liasa (ADSL), se observa el aumento de los metabolitos diagnósticos succinilaminoimidazol carboxamida ribósido (SAICAr) y succiniladenosina (S-ado). C) Cromatogramas y espectros de masas de ácidos orgánicos en orina por GC-MS de una aciduria propiónica, se observan los biomarcadores ácidos 3-hidroxipropiónico (3OHP) y metilcítrico (MC) y la propionilglicina (PG) y el espectro de masas del MC; y de una aciduria isovalérica, se observan los metabolitos marcadores ácidos 2-hidroxiisovalérico (2OHIVA) y 3-hidroxiisovalérico (3OHIVA) e isovalerilglicina (IVG) y el espectro de masas de la IVG. D) Espectros de masas de acilcarnitinas en plasma por ESI-MS/MS de una deficiencia múltiple de acilCoA deshidrogenasa (MADD), se observa el perfil con aumentos de butirilcarnitina (C4), isovalerilcarnitina (C5), hexanoilcarnitina (C6), octanoilcarnitina (C8), hexanoilcarnitina (C6), decanoilcarnitina (C10) y dodecanoilcarnitina (C12); y de una deficiencia de acilCoA deshidrogenasa de cadena media (MCADD), con aumentos de C6, C8 y decenoilcarnitina (C10:1).")
Ejemplos de cromatogramas diagnósticos de diferentes EMH. A) Cromatograma de aminoácidos en plasma por CIO de un paciente de jarabe de arce (MSUD), se observa el aumento de los aminoácidos marcadores de la enfermedad valina (Val), aloisoleucina (aIle), isoleucina (Ile) y leucina (Leu). B) Cromatograma de purinas en líquido cefaloraquídeo (LCR) por HPLC de un paciente con deficiencia en adenilosuccinato liasa (ADSL), se observa el aumento de los metabolitos diagnósticos succinilaminoimidazol carboxamida ribósido (SAICAr) y succiniladenosina (S-ado). C) Cromatogramas y espectros de masas de ácidos orgánicos en orina por GC-MS de una aciduria propiónica, se observan los biomarcadores ácidos 3-hidroxipropiónico (3OHP) y metilcítrico (MC) y la propionilglicina (PG) y el espectro de masas del MC; y de una aciduria isovalérica, se observan los metabolitos marcadores ácidos 2-hidroxiisovalérico (2OHIVA) y 3-hidroxiisovalérico (3OHIVA) e isovalerilglicina (IVG) y el espectro de masas de la IVG. D) Espectros de masas de acilcarnitinas en plasma por ESI-MS/MS de una deficiencia múltiple de acilCoA deshidrogenasa (MADD), se observa el perfil con aumentos de butirilcarnitina (C4), isovalerilcarnitina (C5), hexanoilcarnitina (C6), octanoilcarnitina (C8), hexanoilcarnitina (C6), decanoilcarnitina (C10) y dodecanoilcarnitina (C12); y de una deficiencia de acilCoA deshidrogenasa de cadena media (MCADD), con aumentos de C6, C8 y decenoilcarnitina (C10:1).
La cromatografía de gases (gas chromatography [GC]) es la técnica de excelencia utilizada desde hace décadas para el análisis de ácidos orgánicos, moléculas en general volátiles de bajo peso molecular. En este caso, previamente al análisis en el cromatógrafo, las moléculas se derivatizan (habitualmente se forman trimetilsilil-derivados), y la elución se produce por el flujo de una fase móvil de gas inerte a través de la columna cromatográfica. Históricamente, el detector utilizado era un detector de ionización de llama (flame ionization detector), pero actualmente se utiliza el espectrómetro de masas. La espectrometría de masas (mass espectrometry [MS]) es una técnica analítica en la que los átomos o moléculas de una muestra son ionizados, con mayor frecuencia positivamente, separados por su relación masa/carga (m/z) y posteriormente detectados y registrados. Así, el metabolito al entrar en el espectrómetro se ioniza y se fragmenta según sus características electroquímicas. El conjunto de fragmentos es único y por tanto identificativo de cada metabolito. La cuantificación se realiza por medio de calibradores con estándares internos. Con esta metodología (GC/MS) se identifican y cuantifican los ácidos orgánicos y otros compuestos hidrofóbicos de bajo peso molecular (aminoácidos, ácidos grasos, ácidos biliares….), biomarcadores de numerosísimas enfermedades que afectan al metabolismo intermediario, entre ellas las acidurias orgánicas (fig 1C)9. Para incrementar la sensibilidad de detección se utiliza la monitorización selectiva de iones (selective ion monitoring), procedimiento utilizado, por ejemplo, en la cuantificación de los ácidos grasos de cadena muy larga para el diagnóstico de enfermedades peroxisomales10 (tabla 2).
Aunque las espectrometría de masas aplicada al diagnóstico de EMH se empezó a desarrollar en los años sesenta del siglo XX, ha sido a partir de la década de los 90 cuando hubo un avance extraordinario en el desarrollo de nuevas aplicaciones para el análisis de compuestos polares no volátiles de menor o mayor tamaño de forma rápida, utilizando poca cantidad de muestra e incluso en muestra de sangre y orina en papel, con apenas manipulación previa11. La combinación del sistema de ionización de electrospray (electrospray ionization [ESI]) con la espectrometría de masas en tándem (ESI-MS/MS) es la utilizada para la medida de la concentración de acilcarnitinas (fig. 1D) y otros metabolitos. La separación de isómeros de acilcarnitinas o de ácidos orgánicos requiere una separación previa por cromatografía líquida (HPLC-ESI/MS/MS)12. Otro sistema de ionización utilizado es la ionización química a presión atmosférica (atmospheric pressure chemical ionization [APCI]) que en combinación con la cromatografía líquida (HPLC/APCI/MS/MS) se utiliza para medir el coenzima Q1013. Para el análisis de moléculas más grandes como péptidos, proteínas o glicoproteínas se han desarrollado otras técnicas como, por ejemplo, la que utiliza el sistema de ionización por desorción mediante láser asistida por matriz (matrix-assisted laser desorption/ionization [MALDI]) junto con el analizador de tiempo de vuelo (time of flight [TOF]): MALDI/TOF/MS/MS, en el diagnóstico de hemoglobinopatías14, defectos congénitos de glicosilación15 y diferentes enfermedades de depósito lisosomal16.
Los procedimientos diagnósticos basados en la determinación de metabolitos pueden dar resultados falsos negativos si la muestra no se ha tomado en el momento adecuado (descompensación metabólica), por ello es muy importante conocer cuándo y cómo se ha tomado y conservado la muestra, medicación, dieta, incluyendo suplementos alimenticios y vitaminas que se han administrado al paciente. Los resultados deben ser valorados en laboratorios expertos familiarizados en el estudio y diagnóstico de EMH.
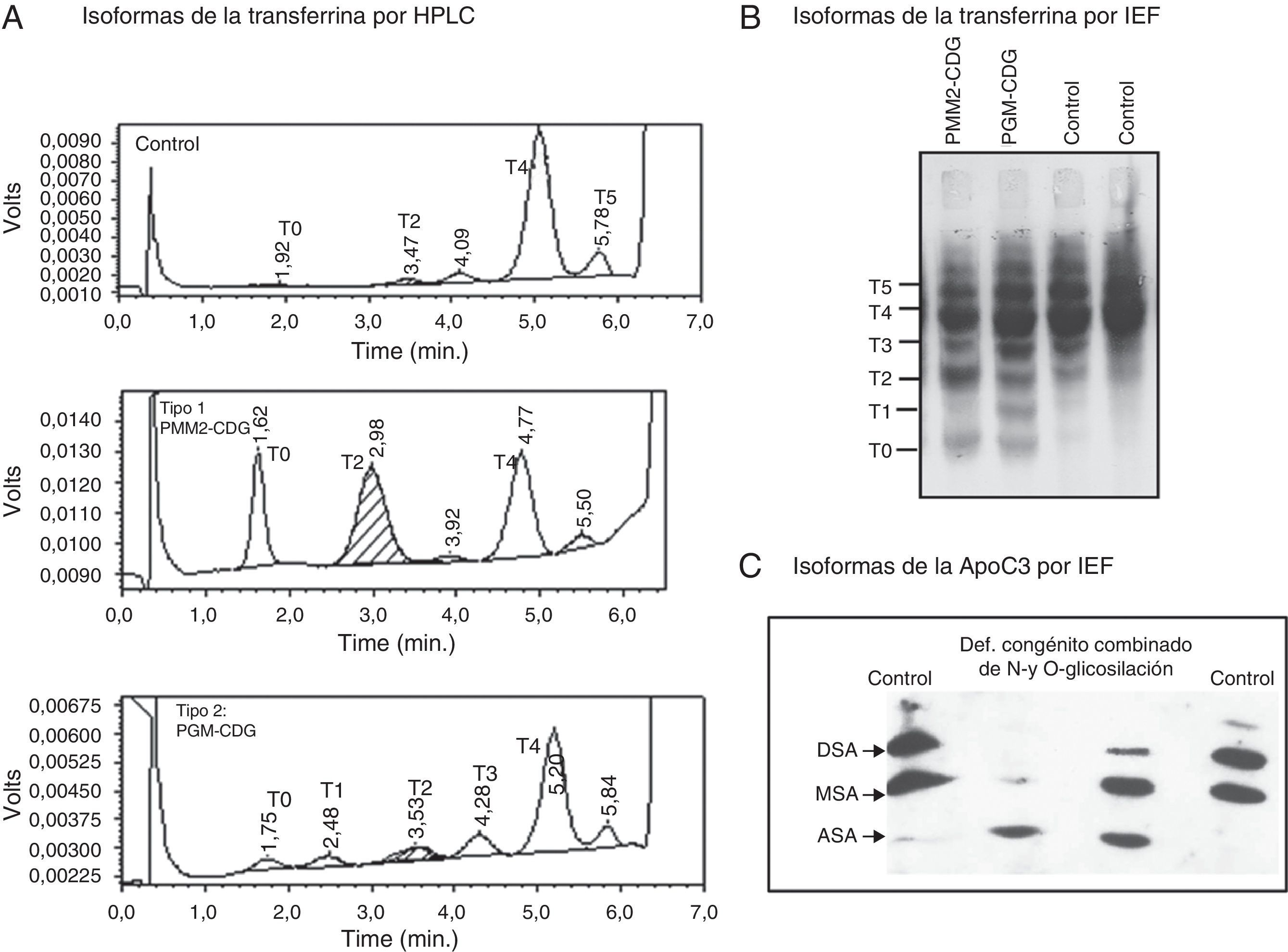
Estudio del producto génico (enzimas y proteínas). El análisis de la proteína afectada a nivel de su función o de su expresión es, en muchos casos, importante bien como confirmación del resultado del análisis de metabolitos o genético o bien cuando no existen metabolitos biomarcadores de la enfermedad. Para el estudio de la función proteica se necesitan células (linfocitos, fibroblastos de piel cultivados a partir de una biopsia de piel, eritrocitos, linfoblastos) o tejidos (biopsia hepática, muscular etc.). La actividad enzimática suele medirse incubando con el sustrato del enzima para medir disminución de sustrato o aparición de producto por técnicas espectrofotométricas o radiactivas. Así se confirma la actividad propionilCoA carboxilasa deficiente en pacientes con aciduria propiónica, utilizando 14CO3H, uno de los sustratos de la carboxilación del metilmalonilCoA17. Las células cultivadas, fundamentalmente fibroblastos de piel y linfoblastos, permiten realizar ensayos in vitro (incorporación de aminoácidos, oxidación de sustratos), para determinar la función de una vía metabólica completa, como por ejemplo la oxidación de 14C-propionato para el análisis de funcionabilidad de la vía del propionato en el diagnóstico de las acidurias metilmalónicas18, de la 14C-leucina en el diagnóstico de la enfermedad jarabe de arce y del 14C-piruvato, para los defectos del complejo piruvato deshidrogenasa y en general, de la función energética mitocondrial. En las enfermedades causadas por un defecto en la glicosilación de proteínas, el análisis en suero de las isoformas de la transferrina, proteína N-glicosilada transportadora de hierro, y de la lipoproteína O-glicosilada ApoC3 se utiliza para orientar junto con la sintomatología clínica el análisis genético. Su concentración en sangre puede ser normal, sin embargo están deficientemente glicosiladas, lo que se traduce en un cambio del perfil de sus isoformas (fig. 2)19.
 Perfil de las isoformas de la transferrina separadas y cuantificadas por HPLC-Variant. La isoforma mayoritaria en suero control es la tetrasialilada (T4); se observa un perfil alterado tipo 1, en un paciente con PMM2-CDG, con aumentos de las isoformas hipoglicosiladas: asialilada (T0) y disialilada (T2) y de tipo 2 en un paciente con PGM-CDG, en donde se observan aumentos de las isoformas: asialilada (T0), monosialilada (T1), disialilada (T2) y trisialilada (T3). En ambos casos se observa la disminución de la isoforma mayoritaria T4. B) Análisis por isoelectroenfoque de la transferrina, en este caso las isoformas se separan por su punto isoeléctrico en gradiente de pH. Se observan las bandas patológicas en los pacientes con PMM2-CDG (T0 y T2) y PGM-CDG (T0, T1, T2 y T3, respectivamente). C) Análisis por isolectroenfoque de la apoC3, las isoformas también se separan en función de su punto isoeléctrico en un gradiente de pH. Los carriles centrales corresponden a dos pacientes con un defecto combinado en la N- y O-glicosilación de proteínas. En un caso se observa aumento de la isoforma asialilada (ASA) y desaparición de las bandas correspondientes a las formas disialilada (DSA) y monosialilada (MSA) y en el otro aumento de la isoforma MSA y fuerte disminución de la DSA de la apoC3.")
Análisis de las isoformas de la N-glicoproteína transferrina y de la
O-glicoproteína lipoproteína apoC3 en suero para la detección de defectos congénitos de glicosilación. A) Perfil de las isoformas de la transferrina separadas y cuantificadas por HPLC-Variant. La isoforma mayoritaria en suero control es la tetrasialilada (T4); se observa un perfil alterado tipo 1, en un paciente con PMM2-CDG, con aumentos de las isoformas hipoglicosiladas: asialilada (T0) y disialilada (T2) y de tipo 2 en un paciente con PGM-CDG, en donde se observan aumentos de las isoformas: asialilada (T0), monosialilada (T1), disialilada (T2) y trisialilada (T3). En ambos casos se observa la disminución de la isoforma mayoritaria T4. B) Análisis por isoelectroenfoque de la transferrina, en este caso las isoformas se separan por su punto isoeléctrico en gradiente de pH. Se observan las bandas patológicas en los pacientes con PMM2-CDG (T0 y T2) y PGM-CDG (T0, T1, T2 y T3, respectivamente). C) Análisis por isolectroenfoque de la apoC3, las isoformas también se separan en función de su punto isoeléctrico en un gradiente de pH. Los carriles centrales corresponden a dos pacientes con un defecto combinado en la N- y O-glicosilación de proteínas. En un caso se observa aumento de la isoforma asialilada (ASA) y desaparición de las bandas correspondientes a las formas disialilada (DSA) y monosialilada (MSA) y en el otro aumento de la isoforma MSA y fuerte disminución de la DSA de la apoC3.
Estudio genético (genes). La confirmación genética de las EMH se lleva a cabo en los laboratorios especializados de genética molecular mediante el análisis de mutaciones del gen correspondiente.
El defecto primario en una EMH es una alteración en la secuencia nucleotídica del ADN que determina la secuencia de aminoácidos de una proteína. La secuencia de nucleótidos del ADN mediante un proceso llamado transcripción genera una cadena de ARN, que se procesa (splicing) para dar lugar al ARN mensajero (ARNm), que se traduce (proceso de traducción) en la secuencia de aminoácidos de la proteína. Finalmente, la proteína es sometida a una serie de modificaciones postraducionales (fosforilación, glicosilación, etc.) para ser plenamente funcional.
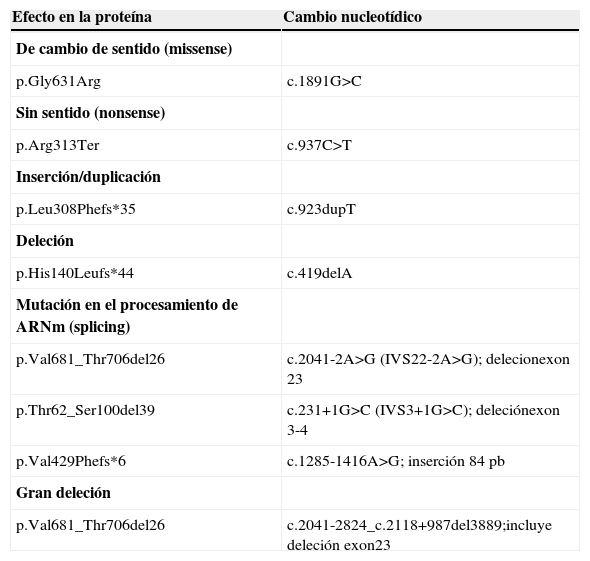
Básicamente, el diagnóstico genético consiste en buscar mutaciones potencialmente patogénicas del gen afectado en material genético (ADN o ARN) del paciente. La muestra a analizar puede ser sangre con anticoagulante (EDTA), sangre seca impregnada en papel de filtro, tejidos como biopsia de hígado o muscular, fibroblastos de piel cultivados, biopsia corial o amniocitos para los estudios prenatales, etc… Las mutaciones pueden afectar a secuencias reguladoras alterando su expresión, secuencias exónicas, alterando la secuencia de aminoácidos de la proteína que codifica, o a regiones intrónicas, que en general afectan al procesamiento (splicing) del ARNm. En la secuencia nucleotídica puede haber mutaciones puntuales que afectan a un solo nucleótido, deleciones o inserciones que afectan a uno o unos pocos nucleótidos y grandes reordenamientos genómicos, es decir deleciones, duplicaciones o inversiones que pueden afectar a uno o más genes. Según su efecto en la proteína, las mutaciones se clasifican como de cambio de sentido (missense) cuando cambia un aminoácido por otro, de cambio sin sentido (nonsense) cuando un aminoácido cambia por un codón de parada de la traducción. En el caso de pequeñas deleciones o inserciones de nucleótidos, estas pueden ser en fase de lectura si la deleción es de tres o múltiplo de tres nucleótidos, eliminándose o insertándose aminoácidos o bien puede producir un cambio en la fase de lectura (frameshift), dando lugar a un codón de parada prematuro (tabla 3). Las mutaciones que afectan al procesamiento del ARNm se denominan mutaciones de splicing, y son causadas por mutaciones puntuales, deleciones o inserciones bien en la secuencia conservada donadora y aceptora de splicing, localizadas en la unión exón-intrón o bien en secuencias exónicas o intrónicas menos conservadas que están implicadas en la regulación del proceso de splicing. El efecto es la generación de transcritos aberrantes portadores de deleciones de secuencias exónicas completas o parciales o inserciones de secuencias intrónicas (pseudogen), transcritos que si son traducidos, dan lugar, en general a proteínas truncadas debido a cambios en la fase de lectura (tabla 3). En la base de datos Human Gene Mutation Database (www.hgmd.org; HGMD) están tabuladas las mutaciones descritas en los genes humanos de las EMH20.
Tipo de mutaciones y nomenclatura
| Efecto en la proteína | Cambio nucleotídico |
|---|---|
| De cambio de sentido (missense) | |
| p.Gly631Arg | c.1891G>C |
| Sin sentido (nonsense) | |
| p.Arg313Ter | c.937C>T |
| Inserción/duplicación | |
| p.Leu308Phefs*35 | c.923dupT |
| Deleción | |
| p.His140Leufs*44 | c.419delA |
| Mutación en el procesamiento de ARNm (splicing) | |
| p.Val681_Thr706del26 | c.2041-2A>G (IVS22-2A>G); delecionexon 23 |
| p.Thr62_Ser100del39 | c.231+1G>C (IVS3+1G>C); deleciónexon 3-4 |
| p.Val429Phefs*6 | c.1285-1416A>G; inserción 84 pb |
| Gran deleción | |
| p.Val681_Thr706del26 | c.2041-2824_c.2118+987del3889;incluye deleción exon23 |
Ejemplos de mutaciones descritas en el gen PCCA (NM_ NM_000282.3), que causan aciduriapropiónica.
El conocimiento de las mutaciones en los afectos de una determinada EMH es de gran utilidad para la búsqueda de una posible correlación genotipo/fenotipo clínico y bioquímico, para establecer la condición de portador de la enfermedad de otros miembros de la familia, para ofrecer un diagnóstico prenatal rápido y fiable y consejo genético; por último, en términos epidemiológicos, para obtener información sobre frecuencia de alelos mutados y su distribución geográfica y poblacional. Además, en los últimos años, el conocimiento de la naturaleza de la mutación y su efecto está permitiendo el desarrollo de tratamientos personalizados basados en el genotipo del paciente21,22.
Técnicas convencionales de diagnóstico genéticoEl desarrollo de la técnica de la reacción en cadena de la polimerasa en 1986 por Kary Mullis23, supuso un hito en el diagnóstico de enfermedades genéticas, ya que mediante esta técnica se amplifica hasta 109 veces una región de copia única del genoma partiendo de ADN genómico (ADNg), obteniéndose así grandes cantidades de la secuencia a analizar. Para el análisis de mutaciones de un gen, básicamente, a partir del ADNg se amplifican en la reacción en cadena de la polimerasa por separado los exones del gen seleccionado junto con sus secuencias intrónicas flanqueantes, que incluyen las secuencias aceptoras y donadoras de splicing o se amplifica el ADN complementario (ADNc), obtenido por retrotranscripción con la enzima transcriptasa en reverso utilizando como sustrato el ARNm. Tras la purificación de los fragmentos obtenidos, se someten a las reacciones de secuenciación mediante el método enzimático de terminación de cadena de ADN por incorporación de dideoxinucleótidos trifosfato (ddNTPs), método de secuenciación de Sanger24, posterior separación en aparatos de electroforesis capilar y detección fluorimétrica (fig. 3A). El análisis partiendo de ARNm es más rápido, ya que de este modo se analizan todos los exones codificantes a la vez en uno o dos fragmentos. Además analizando el ADNc se detectan con mayor facilidad los errores en el procesamiento del ARNm.
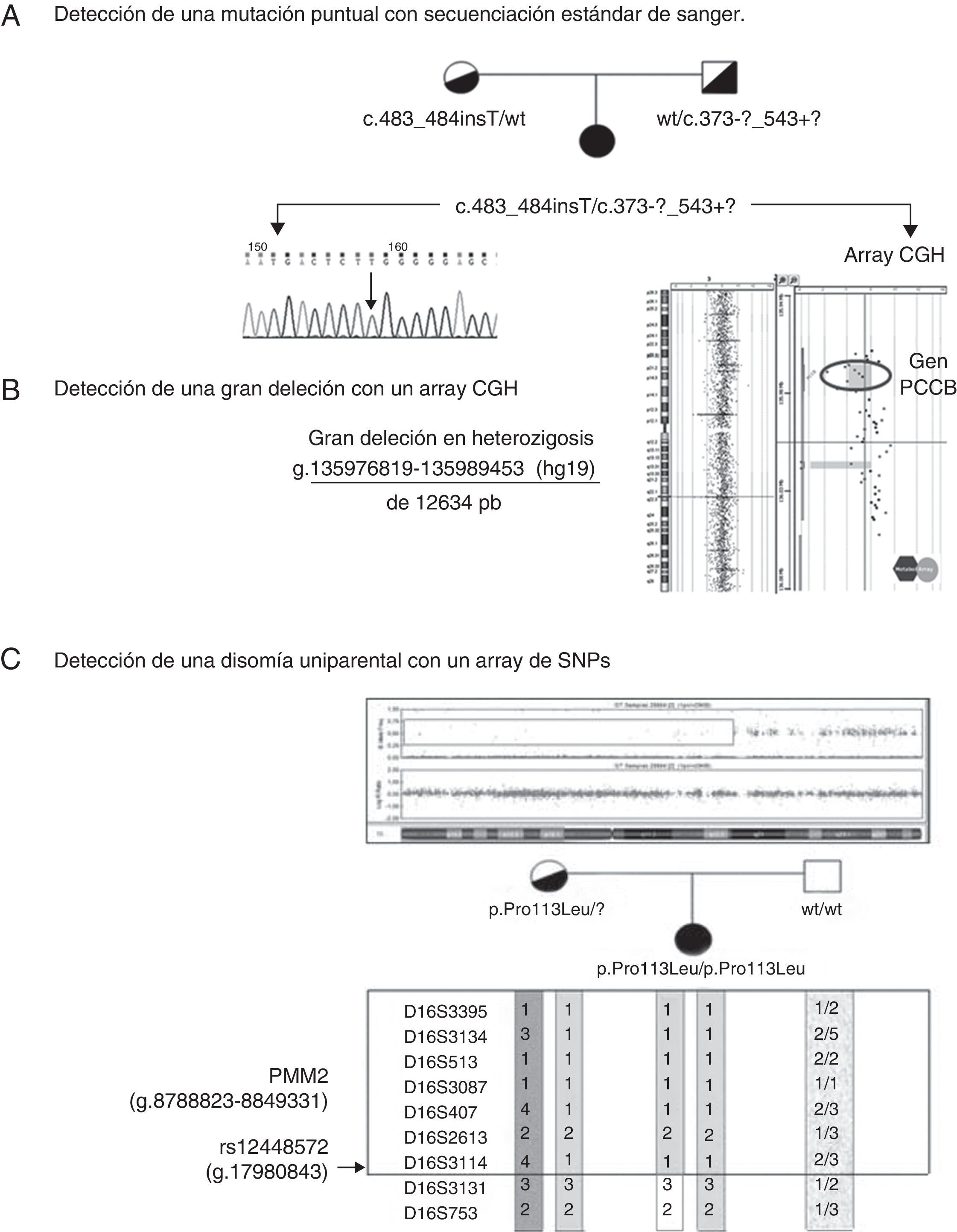
 Secuenciación convencional Sanger del exón 5 del gen PCCB de una muestra de ADN de un paciente con acidemia propiónica. Se observa la inserción de una T entre las posiciones 483 y 484 del cDNA en homocigosis que dará lugar a un cambio en la fase de lectura y a la aparición de un codón de terminación prematura. Esta mutación se encuentra en el alelo materno. B) El análisis de la segregación mendeliana evidenció la posible presencia de una deleción que fue detectada en el alelo paterno mediante hibridación genómica comparada (aCGH metaboloarray®). Se observa una deleción de 12,6 Mb, se indican las coordenadas genómicas, en el cromosoma 3 que incluye parte del gen PCCB. C) Detección de una isodisomía uniparental segmental materna de la región del cromosoma 16 donde se localiza el gen PMM2, en un paciente con un defecto congénito de glicosilación. En el array de SNP se observa la pérdida de heterozigosidad que se confirma mediante el estudio de microsatélites, lo que resulta en la homocigosidad para la mutación patogénica p.Pro113Leu en el caso del índice, siendo el padre no portador de la misma.")
A) Secuenciación convencional Sanger del exón 5 del gen PCCB de una muestra de ADN de un paciente con acidemia propiónica. Se observa la inserción de una T entre las posiciones 483 y 484 del cDNA en homocigosis que dará lugar a un cambio en la fase de lectura y a la aparición de un codón de terminación prematura. Esta mutación se encuentra en el alelo materno. B) El análisis de la segregación mendeliana evidenció la posible presencia de una deleción que fue detectada en el alelo paterno mediante hibridación genómica comparada (aCGH metaboloarray®). Se observa una deleción de 12,6 Mb, se indican las coordenadas genómicas, en el cromosoma 3 que incluye parte del gen PCCB. C) Detección de una isodisomía uniparental segmental materna de la región del cromosoma 16 donde se localiza el gen PMM2, en un paciente con un defecto congénito de glicosilación. En el array de SNP se observa la pérdida de heterozigosidad que se confirma mediante el estudio de microsatélites, lo que resulta en la homocigosidad para la mutación patogénica p.Pro113Leu en el caso del índice, siendo el padre no portador de la misma.
Existen otras técnicas para el análisis de reordenamientos genómicos, que detectan grandes deleciones, inserciones, duplicaciones y disomías uniparentales. La técnica de multiplex-ligation dependent probe amplification, se basa en la hibridación con sondas específicas para los exones del gen que se va a analizar, regiones que posteriormente se ligan, amplifican, se separan por electroforesis capilar y detectan por fluorescencia. Las deleciones se identifican por disminución o ausencia del pico correspondiente al exón o grupo de exones delecionados. Es una técnica complementaria a la secuenciación estándar ya que hay genes en los que se producen deleciones con frecuencia (genes OTC, GLDC, PCCA etc…)25. Las técnicas basadas en arrays son muy versátiles y permiten el análisis de todo el genoma y en varias muestras a la vez. En el caso del array de hibridación genómica comparada (array-CGH), se identifican pérdidas o ganancia de material genético en la muestra de ADN del paciente cuando se compara con una muestra de ADN referencia. Básicamente, se marcan con diferentes fluoróforos el genoma del paciente y del control, se hibrida con el array de oligos de las regiones seleccionadas y se compara la fluorescencia unida a la región seleccionada lo que permite detectar deleciones (pérdida) o duplicaciones (ganancia), (fig. 3B). En los arrays de SNP (polimorfismos de un solo nucleótido), se analizan hasta 106 SNP del genoma; se utilizan en estudios de ligamiento, mapeo de homozigosidad y detección de disomías uniparentales, alteración genética producida cuando se heredan dos cromosomas o segmentos de un cromosoma del mismo progenitor, lo que da lugar a que aparezca una mutación recesiva en homozigosis cuando uno de los progenitores no es portador de la misma (fig. 3C)26.
Por último, existen otras técnicas de rastreo de mutaciones, hoy ya prácticamente en desuso. Las técnicas basadas en el distinto comportamiento electroforético de fragmentos de ADN según su secuencia en geles desnaturalizantes o electroforesis en gel con gradiente desnaturalizante (Denaturing gradient gel elcetroforesis) o en geles no desnaturalizantes (single-stranded conformational polymorphysm), permiten detectar el fragmento de ADN que contiene la mutación. Otro método de rastreo más actual es el high resolution melting, basado en las diferentes temperatura de melting (temperatura a la cual se separan las dos cadenas de ADN), de un fragmento de ADN que contiene una mutación con respecto al fragmento control. Con cualquiera de estas técnicas, se requiere posteriormente la secuenciación estándar (Sanger) del fragmento seleccionado para precisar la mutación específica.
Para un correcto asesoramiento genético en todos los casos es imprescindible el análisis de la segregación mendeliana en muestras de los padres, para confirmar la presencia de las mutaciones en alelos diferentes en el caso de enfermedades de herencia autósomica recesiva, para detectar posibles deleciones o disomías uniparentales en los casos en los que se haya detectado la mutación en homocigosis y para poder identificar mutaciones de novo en enfermedades autosómicas dominantes o ligadas al cromosoma X.
Cribado neonatal de los EMH por tándem masas (MS/MS)El cribado neonatal de enfermedades endocrino-metabólicas es una actividad esencial dentro de la salud púbica y tiene como objetivo la detección presintomática de estas enfermedades mediante pruebas que puedan aplicarse a toda la población de recién nacidos, de manera que la intervención médica a tiempo reduzca la morbilidad, mortalidad y las posibles discapacidades asociadas a dichas enfermedades. Ya hace 50 años se inició el cribado masivo en recién nacidos de la fenilcetonuria o deficiencia del enzima fenilalanina hidroxilasa, que si no es diagnosticada y tratada precozmente, produce retraso mental grave y otras secuelas neurológicas. El método utilizado estaba basado en un sistema de inhibición bacteriana utilizando sangre seca impregnada en papel de filtro que se toma del talón de los recién nacidos27. A lo largo de los años, se han desarrollado diferentes técnicas analíticas para el cribado de diferentes EMH, cromatografía en papel, cromatografía en capa fina, fluorometría, colorimetría, radioinmunoensayo etc. Actualmente, la combinación del sistema de ionización ESI con la espectrometría de masas en tándem (ESI-MS/MS) es la técnica utilizada para la medida de la concentración de aminoácidos y acilcarnitinas en los programas de cribado neonatal ampliados de todo el mundo28. De manera que se detectan un importante número de EMH que afectan al metabolismo de aminoácidos, ácidos orgánicos y de la β-oxidación mitocondrial de ácidos grasos. Los aminoácidos y las acilcarnitinas se extraen de la muestra en papel y se analizan bien derivatizándose mediante butilación (formación de butil-esteres) que favorece la ionización o bien sin derivatizar. La mayoría de los aminoácidos tienen la característica de perder un fragmento neutro de 102Da al chocar con las moléculas del gas en el interior del espectrómetro, generándose un perfil de fragmentos reproducible (NL102). Los aminoácidos básicos solo se pueden analizar mediante el método de «medida de múltiples iones» (multiple reaction monitoring), basado en la selección del fragmento característico de cada uno de ellos. También las acilcarnitinas tienen la característica común de perder un fragmento de masas de 85Da de carga positiva cuando se someten a los choques moleculares dentro del espectrómetro (PS85), obteniéndose un espectro de masas que cubre las acilcarnitinas desde 2 (C2) hasta 20 átomos de carbono (C20). La mayoría de los programas de cribado actuales utilizan la combinación de ensayo de NL102 para aminoácidos, de multiple reaction monitoring para aminoácidos básicos y PS85 para la carnitina y acilcarnitinas de forma secuencial.
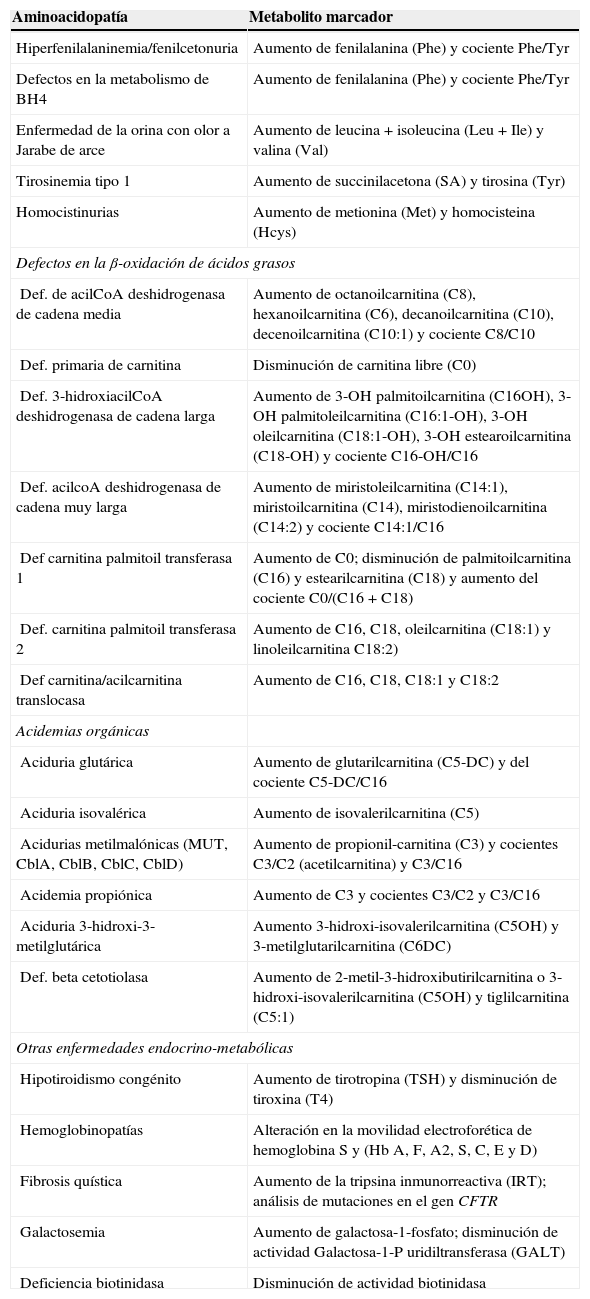
En la actualidad, no existe aún un consenso sobre el número de patologías que deben cribarse. La comisión Europea elaboró un informe publicado en 2012, en el que se pone de manifiesto las diferencias existentes entre países e incluso entre regiones del mismo país29,30. En el año 2006, el Consejo Interterritorial del Sistema Nacional de Salud bajo la coordinación del Ministerio de Sanidad y Asuntos Sociales, elaboró un informe analizando la situación de las actividades del cribado neonatal en las diferentes CC.AA. españolas. En las conclusiones se recomienda una revisión interna de los programas de cribado que garantice el acceso a las mismas detecciones independientemente del lugar de nacimiento del recién nacido (equidad), e insta a la creación de un grupo de trabajo de expertos que definan los criterios a utilizar para incorporar nuevas patologías a cribar y cuáles han de ser estas patologías. Con este motivo, se elaboró un documento en el que se reformulan estos criterios y se hace una valoración de las patologías en base a su importancia en la literatura científica, proponiéndose un panel de 24 enfermedades endocrinas y metabólicas recomendables para cribar (tabla 4)31. El 23 de julio de 2013 se aprobó en el Pleno del Consejo Interterritorial del Sistema Nacional de Salud la relación de enfermedades que formarán parte del programa de cribado neonatal de enfermedades endocrino-metabólicas del Sistema Nacional de Salud: hipotiroidismo congénito, fenilcetonuria, fibrosis quística, deficiencia de acil-CoA deshidrogenasa de cadena media (MCADD), deficiencia de 3-hidroxi acil-CoA deshidrogenasa de cadena larga (LCHADD), acidemia glutárica tipo I (GA-I) y anemia falciforme.
Patologías recomendables para introducir en los programas de cribado neonatal y metabolito marcador
| Aminoacidopatía | Metabolito marcador |
|---|---|
| Hiperfenilalaninemia/fenilcetonuria | Aumento de fenilalanina (Phe) y cociente Phe/Tyr |
| Defectos en la metabolismo de BH4 | Aumento de fenilalanina (Phe) y cociente Phe/Tyr |
| Enfermedad de la orina con olor a Jarabe de arce | Aumento de leucina+isoleucina (Leu+Ile) y valina (Val) |
| Tirosinemia tipo 1 | Aumento de succinilacetona (SA) y tirosina (Tyr) |
| Homocistinurias | Aumento de metionina (Met) y homocisteina (Hcys) |
| Defectos en la β-oxidación de ácidos grasos | |
| Def. de acilCoA deshidrogenasa de cadena media | Aumento de octanoilcarnitina (C8), hexanoilcarnitina (C6), decanoilcarnitina (C10), decenoilcarnitina (C10:1) y cociente C8/C10 |
| Def. primaria de carnitina | Disminución de carnitina libre (C0) |
| Def. 3-hidroxiacilCoA deshidrogenasa de cadena larga | Aumento de 3-OH palmitoilcarnitina (C16OH), 3-OH palmitoleilcarnitina (C16:1-OH), 3-OH oleilcarnitina (C18:1-OH), 3-OH estearoilcarnitina (C18-OH) y cociente C16-OH/C16 |
| Def. acilcoA deshidrogenasa de cadena muy larga | Aumento de miristoleilcarnitina (C14:1), miristoilcarnitina (C14), miristodienoilcarnitina (C14:2) y cociente C14:1/C16 |
| Def carnitina palmitoil transferasa 1 | Aumento de C0; disminución de palmitoilcarnitina (C16) y estearilcarnitina (C18) y aumento del cociente C0/(C16+C18) |
| Def. carnitina palmitoil transferasa 2 | Aumento de C16, C18, oleilcarnitina (C18:1) y linoleilcarnitina C18:2) |
| Def carnitina/acilcarnitina translocasa | Aumento de C16, C18, C18:1 y C18:2 |
| Acidemias orgánicas | |
| Aciduria glutárica | Aumento de glutarilcarnitina (C5-DC) y del cociente C5-DC/C16 |
| Aciduria isovalérica | Aumento de isovalerilcarnitina (C5) |
| Acidurias metilmalónicas (MUT, CblA, CblB, CblC, CblD) | Aumento de propionil-carnitina (C3) y cocientes C3/C2 (acetilcarnitina) y C3/C16 |
| Acidemia propiónica | Aumento de C3 y cocientes C3/C2 y C3/C16 |
| Aciduria 3-hidroxi-3-metilglutárica | Aumento 3-hidroxi-isovalerilcarnitina (C5OH) y 3-metilglutarilcarnitina (C6DC) |
| Def. beta cetotiolasa | Aumento de 2-metil-3-hidroxibutirilcarnitina o 3-hidroxi-isovalerilcarnitina (C5OH) y tiglilcarnitina (C5:1) |
| Otras enfermedades endocrino-metabólicas | |
| Hipotiroidismo congénito | Aumento de tirotropina (TSH) y disminución de tiroxina (T4) |
| Hemoglobinopatías | Alteración en la movilidad electroforética de hemoglobina S y (Hb A, F, A2, S, C, E y D) |
| Fibrosis quística | Aumento de la tripsina inmunorreactiva (IRT); análisis de mutaciones en el gen CFTR |
| Galactosemia | Aumento de galactosa-1-fosfato; disminución de actividad Galactosa-1-P uridiltransferasa (GALT) |
| Deficiencia biotinidasa | Disminución de actividad biotinidasa |
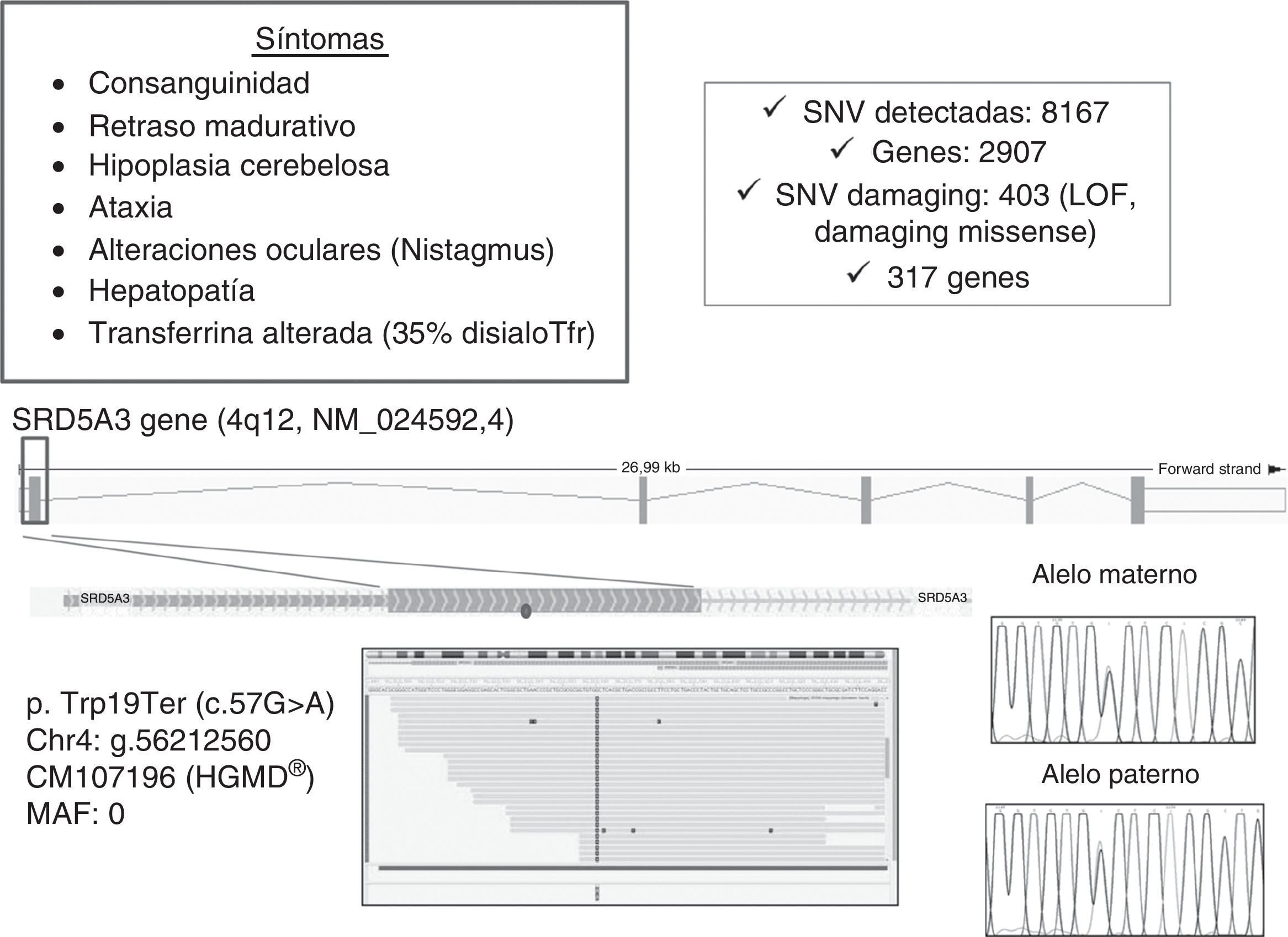
La tecnología de la secuenciación masiva del ADN con los equipos de secuenciación de nueva generación (next generation sequencing) permite la secuenciación del genoma completo o del exoma celular (suma de los exones o regiones codificantes más regiones intrónicas adyacentes) de forma cada vez más rápida y eficaz y está suponiendo un avance sin precedentes en la elucidación de las bases moleculares de miles de enfermedades genéticas, entre ellas las EMH. En el mercado existen diferentes instrumentos para la secuenciación masiva, pero para todos se utiliza un esquema de trabajo similar. Se requiere la fragmentación del ADN y amplificación clonal masiva de los fragmentos generados seguida de bien pirosecuenciación (plataforma Roche 454/FLX), bien secuenciación por síntesis (plataforma Illumina) o bien secuenciación en una reacción de ligación (plataforma SOLID)32. Tras la generación de librerías clonales, se alinean las miles de secuencias generadas con la secuencia del genoma humano de referencia. La gran cantidad de variantes detectadas se filtran por una serie de programas informáticos hasta determinar cuáles podrían ser las variantes causantes de enfermedad. Básicamente, se analiza su presencia o no en bases de datos de polimorfismos (dbSNP, db 1000 genomas, EVS, db del laboratorio) y de mutaciones ya descritas [HMGD]), se analiza su efecto funcional sobre la proteína, siendo buenas candidatas las variantes de cambio de sentido, pequeñas duplicaciones o inserciones que afectan a la fase de lectura (generan frameshifts o codones de parada de la traducción) o que afectan a las regiones conservadas del procesamiento del ARNm. Las variantes de cambio aminoacídico (missense) se evalúan mediante análisis estructural en programas bioinformáticos (SIFT,polyphen2 etc.), que predicen su efecto patogénico sobre la proteína (fig. 4). En todos los casos, las mutaciones se confirman analizando la segregación mendeliana en ADN de los padres mediante la secuenciación de Sanger. La secuenciación masiva del exoma se está utilizando en la práctica clínica, en algunos casos mediante la estrategia de paneles para grupos de EMH clínicamente definidos pero con gran heterogeneidad genética, como por ejemplo las enfermedades mitocondriales33 o los defectos congénitos de glicosilación34.
 patogénicas, de las cuales una (c.57G>A) se encontraba en la región codificante del gen SRD5A3 en homocigosis. Este cambio tiene un efecto presumiblemente severo, ya que da lugar a un codón de parada prematuro (p.Trp19Ter). La presencia de la mutación en homocigosis se confirmó en muestras de los padres por secuenciación Sanger.")
Estudio genético en un caso con sospecha clínica y bioquímica de un defecto congénito de glicosilación. Se analizó el ADN del paciente utilizando el panel de genes TrusightOne, para el análisis del exoma de 4800 genes del OMIM relacionados con enfermedad. Tras la captura y secuenciación de los fragmentos en un Myseq, se alinearon las secuencias y se detectaron 403 variaciones de un solo nucleótido (SNV-single nucleotide variation) patogénicas, de las cuales una (c.57G>A) se encontraba en la región codificante del gen SRD5A3 en homocigosis. Este cambio tiene un efecto presumiblemente severo, ya que da lugar a un codón de parada prematuro (p.Trp19Ter). La presencia de la mutación en homocigosis se confirmó en muestras de los padres por secuenciación Sanger.
El ESHG Quality Committee ha publicado muy recientemente unas recomendaciones para la elaboración de informes en un intento de armonizar su presentación en Europa y países vecinos35. El informe es un documento médico emitido desde el laboratorio al clínico que envía las muestras en las que se refieren los resultados interpretados de las pruebas bioquímicas y genéticas realizadas al paciente. Debe ser claro, conciso, preciso e interpretativo, con el objetivo último de proporcionar respuestas útiles para el diagnóstico del paciente.
ConclusiónLa implementación de los dos avances tecnológicos más interesantes de los últimos años, la espectrometría de masas para el análisis de metabolitos y proteínas, y la secuenciación masiva en el análisis del ADN, han tenido y van a seguir teniendo un gran impacto sobre el modo de diagnosticar las EMH. Tanto para el cribado de población asintomática (neonatal) como para el de la sintomática, la espectrometría de masas tiene un gran potencial en el desarrollo de métodos muy sensibles para la identificación y cuantificación de metabolitos (metaboloma)36 o proteínas (proteoma)37 en muestras humanas no invasivas38. Por otro lado, los métodos para la secuenciación masiva del genoma y del exoma son cada vez más económicos y rápidos en la obtención de resultados y parece inevitable en un futuro no muy lejano su aplicación en el diagnóstico prenatal39 o en el cribado de población asintomática. Aunque en este caso, todavía quedan numerosas cuestiones prácticas y éticas que resolver, especialmente las que se derivan de la necesidad de la aceptación por parte del paciente o de sus padres de un consentimiento informado complejo, y del manejo de numerosos datos genéticos, algunos de ellos de significado incierto40. La identificación de portadores de enfermedades graves y la detección en un recién nacido de mutaciones relacionadas con enfermedad o riesgo de enfermedad de comienzo en la edad adulta requerirán un buen asesoramiento genético41.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a todo el personal científico, técnico y administrativo del Centro de Diagnóstico de Enfermedades Moleculares de la Universidad Autónoma de Madrid, su excelente trabajo durante estos años, muy especialmente a su directora, la Profesora Magdalena Ugarte. También agradecen a Rosa Navarrete la composición de las figuras de este trabajo.
