


La fibrosis quística es la enfermedad de herencia autosómica recesiva grave más frecuente en población caucásica. Su prevalencia en los países de Europa occidental es de un caso de cada 2.000 a 6.000 recién nacidos vivos. Los pacientes desarrollan una enfermedad pulmonar crónica y progresiva, que es la causa más frecuente de la morbimortalidad. En el 85% de los casos existe, además, insuficiencia pancreática. Son frecuentes las alteraciones hepatobiliares y genitourinarias, la azoospermia, aunque la enfermedad pulmonar y la insuficiencia pancreática son las que determinan la gravedad del proceso. Es una enfermedad genética causada por defectos en el gen CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), en el que se han descrito hasta la fecha más de 1.900 mutaciones. El diagnóstico de la enfermedad es fundamentalmente clínico y la confirmación se realiza mediante la detección de niveles altos de cloro en sudor y la identificación de mutaciones en el gen CFTR.
Cystic fibrosis is the most severe common autosomal recessive disease in caucasian population. Its prevalence in countries of Western Europe is one case in 2.000-6.000 live births. Patients develop a chronic, progressive lung disease, which is the most common cause of morbidity and mortality. In 85% of cases there is also pancreatic insufficiency. Hepatobiliary and genitourinary disorders and azoospermia are frequent, although lung symptoms and pancreatic insufficiency determine the severity of the disease. It is a genetic disease caused by defects in the CFTR gene (Cystic Fibrosis Transmembrane Conductance Regulator). To date more than 1900 mutations have been reported. Diagnosis of the disease is essentially clinical and confirmation is made by the detection of high levels of sweat chloride and the identification of mutations in the CFTR gene.
La fibrosis quística (FQ), descrita en 1938 como «fibrosis quística del páncreas» y posteriormente denominada «mucoviscidosis», es una de las enfermedades de herencia autosómica recesiva más frecuentes en la raza caucásica, con una incidencia estimada entre 1/2.000 y 1/6.000 nacidos vivos. En España disponemos de datos obtenidos por algunas Comunidades Autónomas que tienen desde hace años esta enfermedad en su programa de cribado neonatal y la incidencia detectada oscila entre 1/4.500 en Castilla y León1 hasta 1/6.5002 en Cataluña.
La enfermedad se caracteriza por una acumulación excesiva de moco espeso y viscoso en el epitelio del sistema respiratorio y del tracto digestivo. Casi todos los pacientes desarrollan una enfermedad crónica y progresiva del aparato respiratorio, que es la causa más frecuente de la morbimortalidad. En el 85% de los casos existe disfunción pancreática (exocrina o endocrina). Aunque también son frecuentes las alteraciones hepatobiliares y genitourinarias, con azoospermia obstructiva en los varones, la insuficiencia pancreática y la enfermedad pulmonar son las que determinan la gravedad del proceso así como su pronóstico3.
La FQ es una enfermedad genética causada por defectos en el gen regulador de la conductabilidad transmembrana (Cystic Fibrosis Transmembrane Conductance Regulator –CFTR–), ubicado en el brazo largo del cromosoma 7. El gen codifica una proteína transmembrana de 1.480 aminoácidos que es un canal de iones cloruro (Cl−) y que, además, actúa como regulador de diversos canales iónicos.
Durante los 30 últimos años se ha duplicado la esperanza de vida de las personas con FQ. En los años 90, en el Reino Unido, se pronosticó que la esperanza de vida media para una cohorte de lactantes con FQ sería de alrededor de los 40 años4. Gran parte de este aumento se atribuyó a las mejoras en el tratamiento. y se argumentó que las intervenciones terapéuticas administradas antes de la aparición de los signos o los síntomas podían presentar un beneficio mayor a largo plazo. Como resultado surgió la hipótesis de que el diagnóstico presintomático, mediante el cribado neonatal y la aplicación de tratamiento precoz, podía prevenir o reducir el daño pulmonar irreversible y optimizar el estado nutricional, mejorando así la calidad de vida en las personas con FQ.
La FQ constituye un problema de salud importante y, en opinión de la mayoría de los autores, cumple desde hace años los criterios generalmente aceptados para ser incluida en los programas de cribado neonatal5; ya que es una enfermedad bien definida clínica y bioquímicamente, con morbimortalidad importante, tiene un tratamiento eficaz, aunque no curativo, que mejora claramente su evolución. Existe además un marcador bioquímico, la tripsina inmunorreactiva en sangre (TIR), de bajo coste y sensibilidad y especificidad aceptables. Por ello, en la actualidad todas las Comunidades Autónomas tienen incluida esta enfermedad en sus programas de cribado neonatal siendo una de las siete enfermedades incluidas en la cartera básica común de servicios del Sistema Nacional de Salud.
FisiopatologíaLa alteración del transporte de electrolitos, particularmente de iones cloruro, es la principal anomalía en la FQ. En 1948, el Dr. Paul di Sant Agnese, descubrió la excesiva concentración de sal en el sudor de varios niños afectados de FQ. Con este hallazgo contribuyó a que Gibson y Cooke6 diseñaran un test para medir, de forma estandarizada, la concentración de iones en el sudor, prueba que en la actualidad continúa siendo la de referencia en el diagnóstico de la FQ.
En los años 80, Knowles7 demostró que la anomalía en la composición del sudor se debe a un defecto en la secreción del cloro, activada por AMPc, a nivel del conducto excretor de la glándula sudorípara. El flujo alterado de iones cloruro afecta además al flujo de los iones sodio y el agua que les acompaña, provocando como resultado final la formación de secreciones anormalmente espesas y deshidratadas en diversos órganos. La formación de estas secreciones da lugar a la obstrucción de los conductos del páncreas, glándulas salivares, epidídimo, intestino, bronquios y bronquiolos.
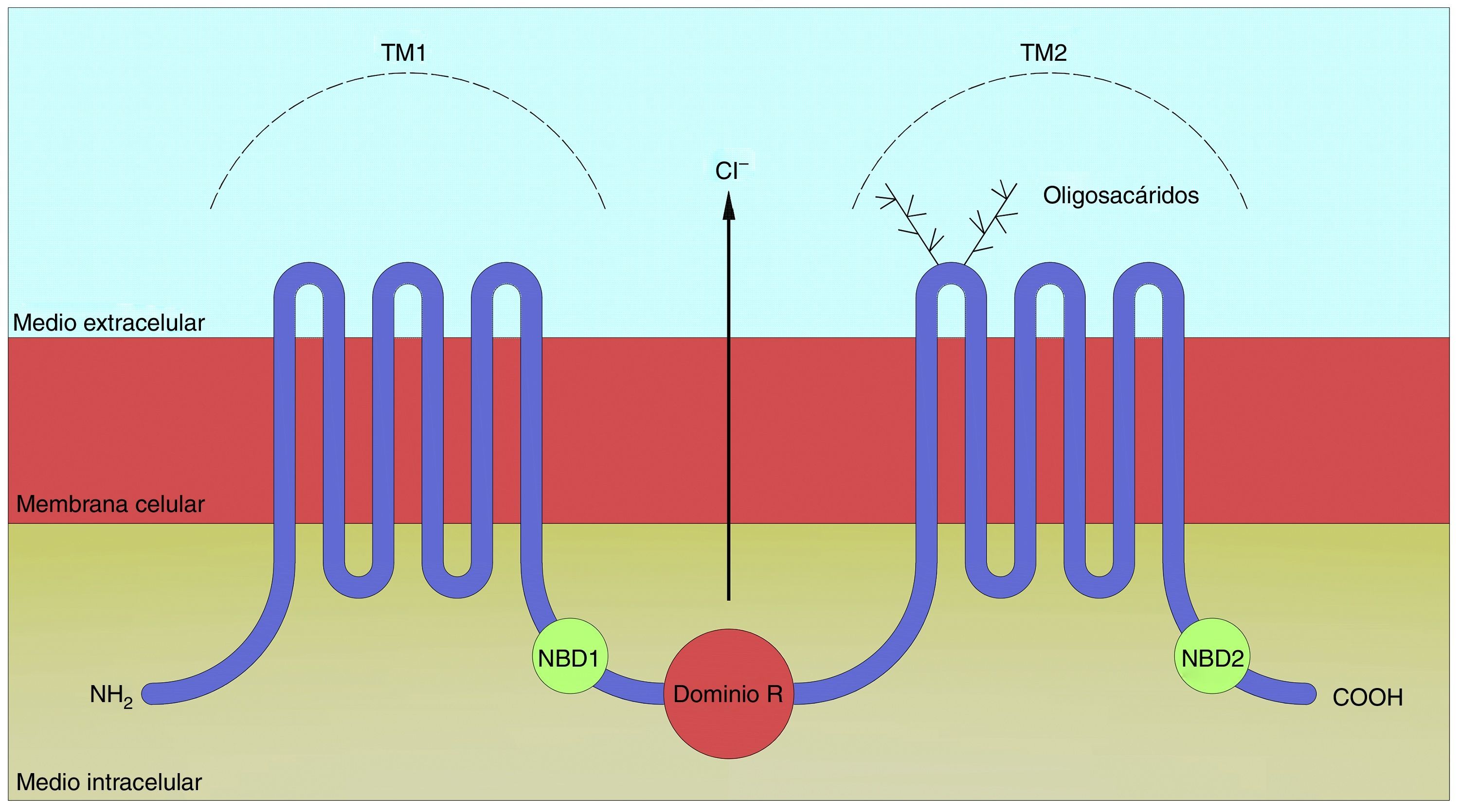
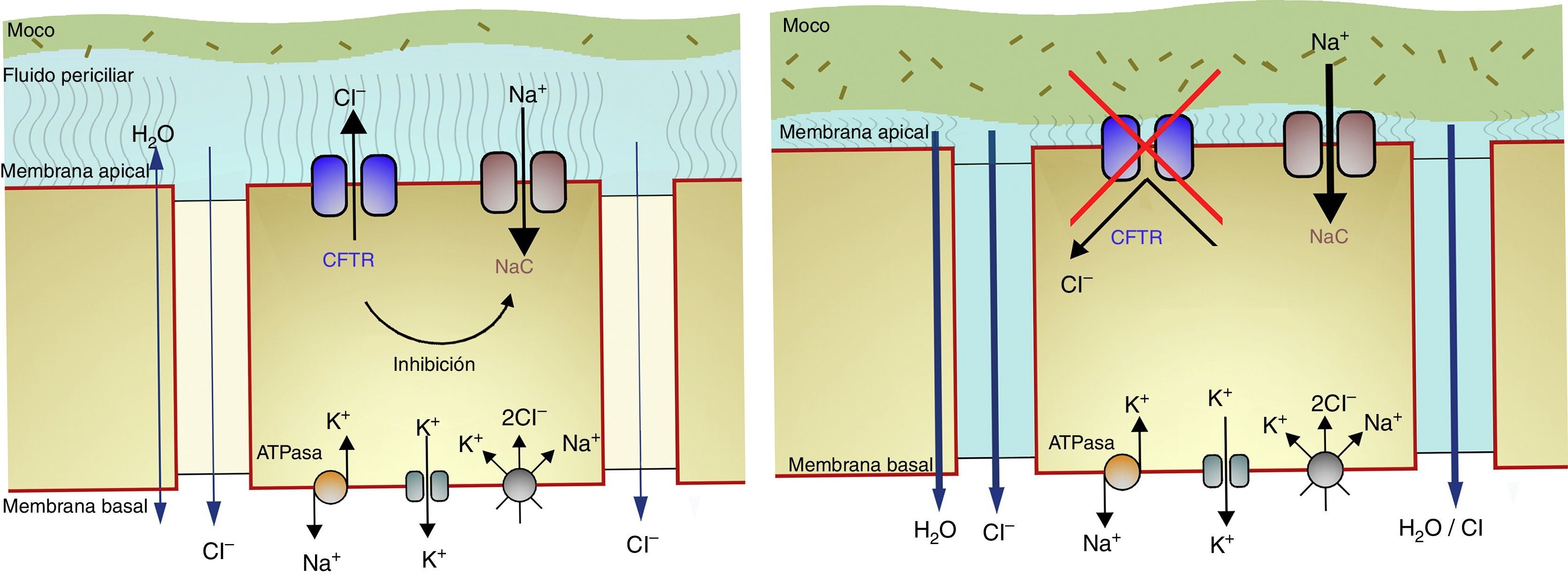
A través de la membrana basal de la célula epitelial de las vías respiratorias de un individuo sano, se produce la entrada de iones sodio y agua de forma pasiva y de iones cloruro en contra de gradiente electroquímico, gracias a que es cotransportado a través de la bomba sodio-potasio (fig. 1). En la membrana apical de la célula epitelial, los iones cloruro salen a favor de gradiente cuando se abren los canales de cloro. Simultáneamente, sale agua, que contribuye a mantener la fluidez de la secreción. En la misma membrana apical existen también canales para el sodio, que permiten su entrada a favor de gradiente electroquímico y que se cierran cuando se produce la secreción de cloro.
 NBD1 y NBD2 y el dominio de regulación R.")
En la FQ el canal CFTR, o está ausente o no responde a los estímulos hormonales fisiológicos, de modo que el flujo de cloro queda bloqueado y con ello también se reduce el flujo de agua al exterior. Paralelamente, aumenta la reabsorción de sodio (y agua) a través de la membrana apical. El resultado final es que la secreción se deshidrata.
Actualmente se acepta que son las secreciones espesas las causantes de la insuficiencia pancreática, de la infertilidad del 90% de los varones enfermos, de las obstrucciones de los bronquios y las infecciones respiratorias crónicas8,9.
Esta alteración a nivel pulmonar origina un aumento de la viscosidad de las secreciones, obstrucción de las vías aéreas y alteración del aclaramiento mucociliar, facilitando la colonización bacteriana y liberación de mediadores proinflamatorios en las vías aéreas. Estos pacientes son susceptibles de desarrollar infección crónica por una serie de patógenos característicos, siendo el más importante de ellos la Pseudomonas aeruginosa (P. aeruginosa). La afectación progresiva del aparato respiratorio dará lugar a la aparición de bronquiectasias, destrucción del parénquima pulmonar, aparición de insuficiencia respiratoria progresiva, hipertensión pulmonar y, en última instancia, la muerte.
GenéticaLa FQ (MIM#219700) es una enfermedad causada por mutaciones en el gen CFTR10,11, en el que se han descrito hasta la fecha más de 1.900 variantes.
Patrón de herencia de la FQEs una enfermedad monogénica con herencia autosómica recesiva. Se manifiesta cuando se han heredado dos copias mutadas del gen CFTR (una de cada progenitor). Los individuos que tienen una copia normal y otra mutada, son portadores de la enfermedad. En el caso de que ambos progenitores sean portadores sanos, existe un 75% de probabilidades de que los hijos sean sanos (un 50% portadores) y un 25% de que estén afectados de FQ.
El gen CFTREl gen CFTR (MIM# 602421) se localiza en la posición q31.2 del brazo largo del cromosoma 710,12. Se extiende a lo largo de 250kb y da lugar a un tránscrito que mide 6.100 nucleótidos de los cuales 4.440 (distribuidos en 27 exones) son la secuencia codificante de la proteína CFTR. La información de ese RNA es traducida a una glicoproteína transmembrana de 1.480 aminoácidos con un peso molecular de aproximadamente 170kDa.
Se expresa en células epiteliales de tejidos exocrinos, como pulmones, páncreas, glándulas sudoríparas, intestino, epidídimo y conductos deferentes.
La proteína CFTR tiene dos dominios transmembrana, cada uno de ellos formado por seis regiones hidrófobas que están ancladas en la bicapa lipídica de la membrana celular (fig. 1). Tienen 2 sitios de unión a ATP y un dominio regulador (dominio R), que contiene múltiples lugares para la fosforilación por una proteína quinasa A. Su función principal es la de actuar como canal de cloro estimulado por AMPc13, además de influir en la actividad de otros canales y en la regulación de otras proteínas (fig. 2).
 y del defecto de la secreción de iones cloro con aumento de reabsorción de sodio y de agua en el epitelio respiratorio de un paciente con FQ (imagen de la derecha).")
El transporte de electrolitos en el epitelio de las vías respiratorias. Esquema del canal CFTR en una persona sana (imagen de la izquierda) y del defecto de la secreción de iones cloro con aumento de reabsorción de sodio y de agua en el epitelio respiratorio de un paciente con FQ (imagen de la derecha).
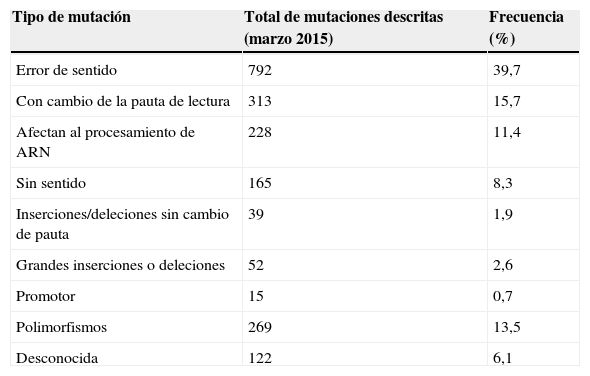
Hasta la fecha, se han descrito cerca de 2.000 mutaciones del gen CFTR que afectan de distinta forma a la estructura y la funcionalidad de la proteína, reduciendo o incluso impidiendo el transporte de electrolitos a través de la membrana de las células epiteliales de los tejidos en los que se expresa este gen. Estas mutaciones están recogidas en la Cystic Fibrosis Mutation Batabase (CFMDB, http://www.genet.sickkids.on.ca/app). Las mutaciones se distribuyen por todo el gen CFTR y la mayoría de las causantes de la enfermedad son mutaciones puntuales en las que solo se ve afectado un nucleótido. De las mutaciones encontradas se estima que un 40,2% son missense (mutaciones donde el cambio de base produce un codón que codifica otro aminoácido); un 15,9% son mutaciones frameshift (desplazamiento en el marco de lectura); un 11,7% son mutaciones de splicing (mutaciones que afectan al procesamiento del ARN); un 8,3% son mutaciones nonsense (mutaciones que dan lugar a un codón de terminación); y un 2,0% son deleciones/inserciones con o sin cambio de pauta de lectura. Además, un 14,3% de las variantes descritas son polimorfismos, mientras que se desconoce las consecuencias funcionales del 3,0% de las mismas (tabla 1).
Distribución de los tipos de mutaciones en el gen CFTR
| Tipo de mutación | Total de mutaciones descritas (marzo 2015) | Frecuencia (%) |
|---|---|---|
| Error de sentido | 792 | 39,7 |
| Con cambio de la pauta de lectura | 313 | 15,7 |
| Afectan al procesamiento de ARN | 228 | 11,4 |
| Sin sentido | 165 | 8,3 |
| Inserciones/deleciones sin cambio de pauta | 39 | 1,9 |
| Grandes inserciones o deleciones | 52 | 2,6 |
| Promotor | 15 | 0,7 |
| Polimorfismos | 269 | 13,5 |
| Desconocida | 122 | 6,1 |
Fuente: Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca/app).
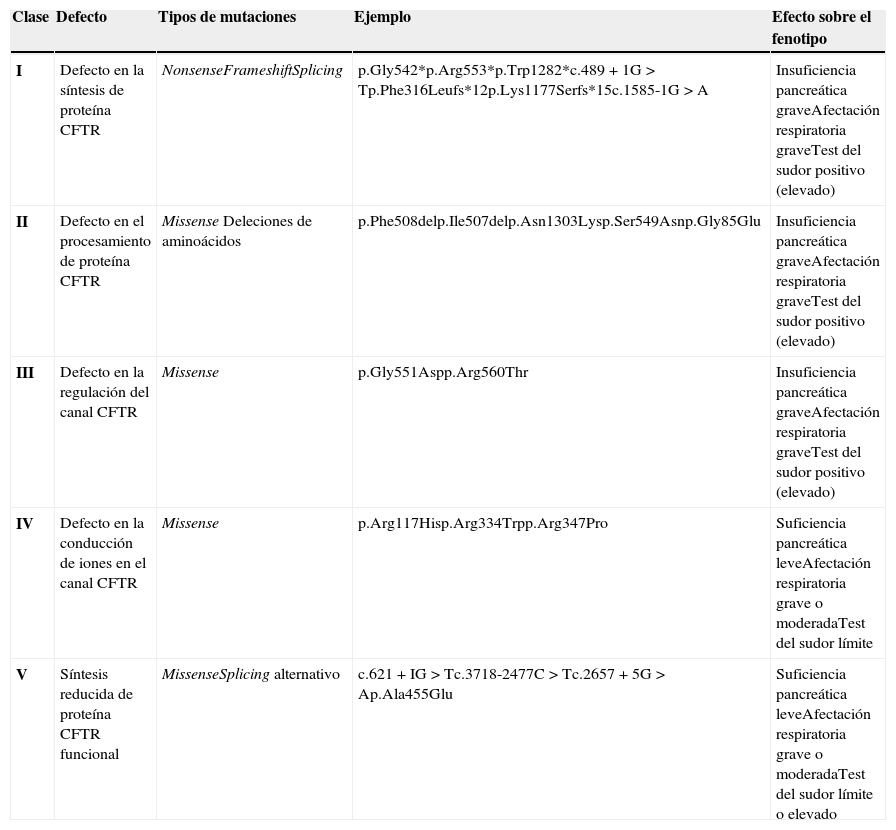
Dependiendo del efecto a nivel de la proteína, las mutaciones del gen CFTR pueden dividirse en cinco clases14:
- -
Clase I: mutaciones que provocan la ausencia total de proteína CFTR. Son mutaciones nonsense, frameshift o de splicing, que producen proteínas truncadas o transcritos inestables que son rápidamente degradados.
- -
Clase II: mutaciones que generan proteínas que no se pliegan adecuadamente, no consiguen madurar y son degradadas sin alcanzar la membrana plasmática.
- -
Clase III: mutaciones que alteran la regulación (apertura/cierre) del canal CFTR. Es el caso de algunas mutaciones missense, que afectan a la unión de ATP en el dominio regulador.
- -
Clase IV: mutaciones que provocan una conducción anómala del flujo de iones cloruro a través del canal CFTR. Estas mutaciones son de tipo missense y se localizan en los dominios transmembrana.
- -
Clase V: mutaciones que producen como resultado cantidades residuales de proteína CFTR funcional.
Las mutaciones de clase I, II y III se consideran mutaciones graves y cursan con insuficiencia pancreática, mientras que las mutaciones de clase IV y V son mutaciones más leves, que suelen asociarse en parte con la mejor función pulmonar, estado nutricional, suficiencia pancreática y colonización por Pseudomonas más tardía (tabla 2). En un paciente con dos mutaciones en el gen CFTR primará el fenotipo causado por la mutación más leve.
Correlación genotipo-fenotipo
| Clase | Defecto | Tipos de mutaciones | Ejemplo | Efecto sobre el fenotipo |
|---|---|---|---|---|
| I | Defecto en la síntesis de proteína CFTR | NonsenseFrameshiftSplicing | p.Gly542*p.Arg553*p.Trp1282*c.489+1G>Tp.Phe316Leufs*12p.Lys1177Serfs*15c.1585-1G>A | Insuficiencia pancreática graveAfectación respiratoria graveTest del sudor positivo (elevado) |
| II | Defecto en el procesamiento de proteína CFTR | Missense Deleciones de aminoácidos | p.Phe508delp.Ile507delp.Asn1303Lysp.Ser549Asnp.Gly85Glu | Insuficiencia pancreática graveAfectación respiratoria graveTest del sudor positivo (elevado) |
| III | Defecto en la regulación del canal CFTR | Missense | p.Gly551Aspp.Arg560Thr | Insuficiencia pancreática graveAfectación respiratoria graveTest del sudor positivo (elevado) |
| IV | Defecto en la conducción de iones en el canal CFTR | Missense | p.Arg117Hisp.Arg334Trpp.Arg347Pro | Suficiencia pancreática leveAfectación respiratoria grave o moderadaTest del sudor límite |
| V | Síntesis reducida de proteína CFTR funcional | MissenseSplicing alternativo | c.621+IG>Tc.3718-2477C>Tc.2657+5G>Ap.Ala455Glu | Suficiencia pancreática leveAfectación respiratoria grave o moderadaTest del sudor límite o elevado |
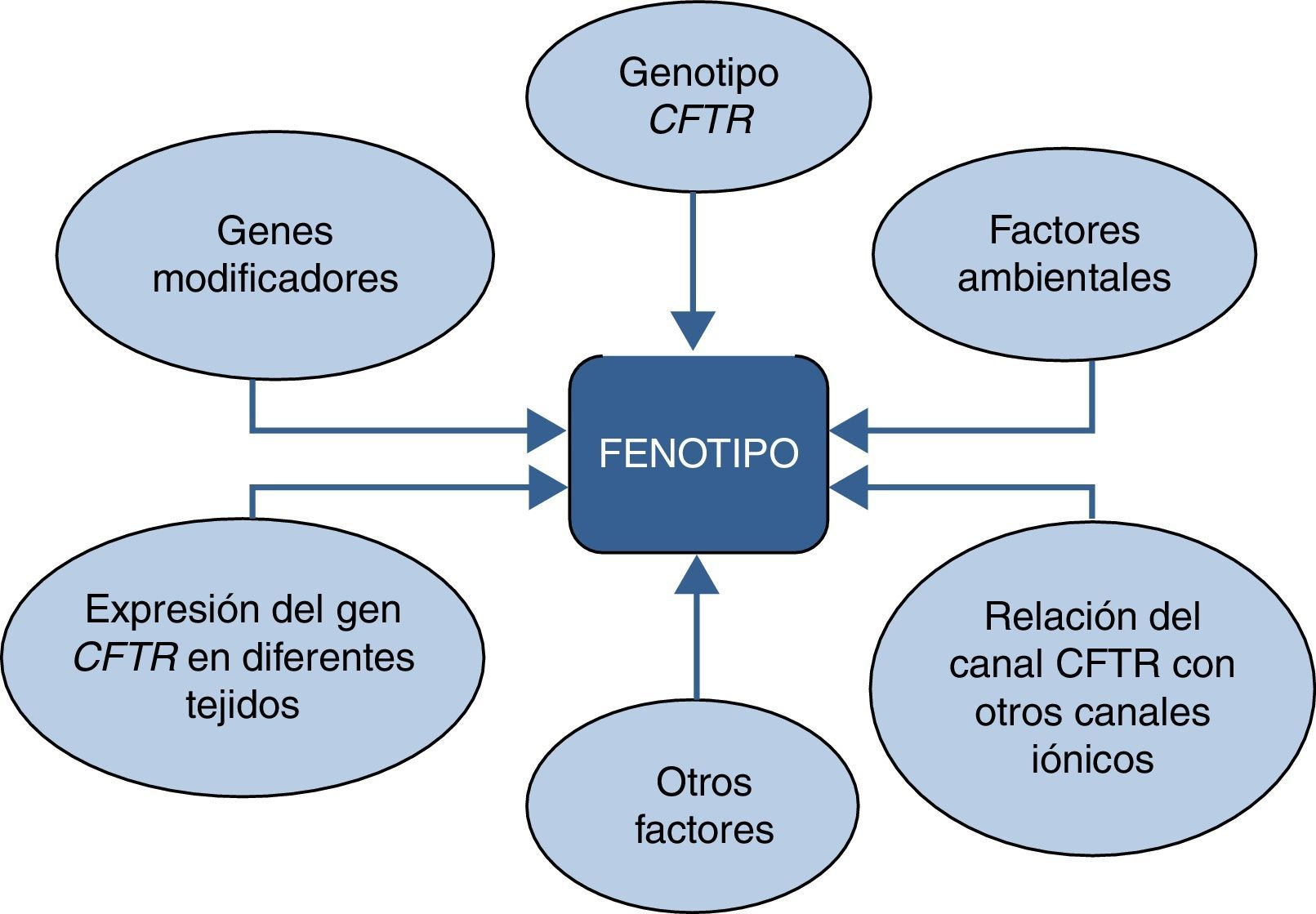
Ciertos genotipos FQ están relacionados con formas clínicas más benignas, pero el valor pronóstico del genotipo FQ respecto a la supervivencia no ha sido todavía claramente establecido. El fenotipo en un individuo afecto de FQ resulta de la interacción compleja entre el genotipo CFTR, la influencia de genes modificadores y la exposición a diferentes agentes ambientales. Es por ello que la clasificación mencionada ofrece solamente información para grupos de enfermos y no a nivel individual, donde solo proporciona información de riesgo (fig. 3).
Distribución de las mutaciones en CFTR
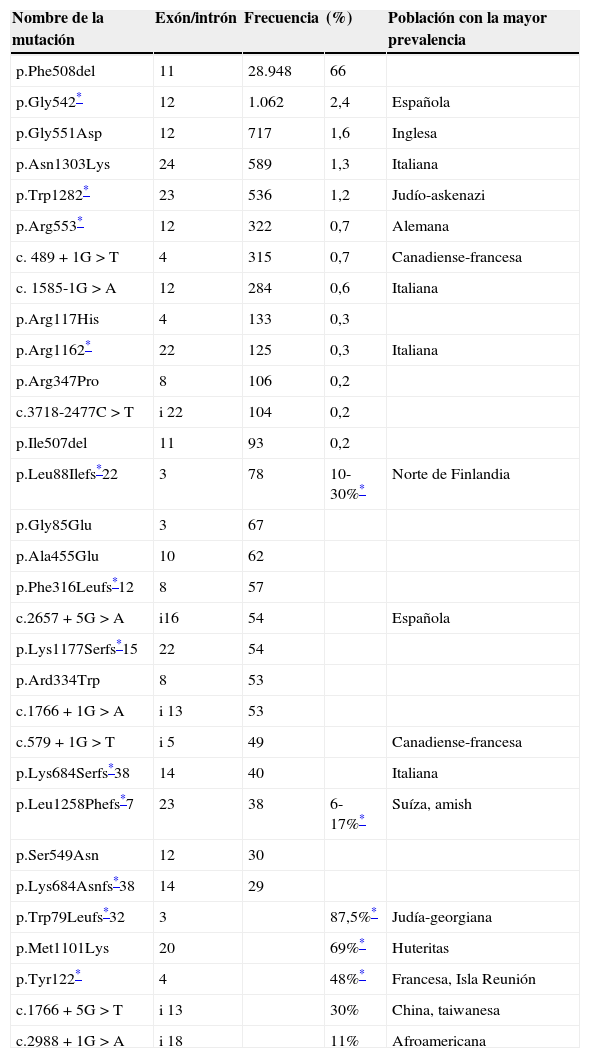
La distribución de las mutaciones en el gen CFTR difiere entre las diferentes poblaciones. La mutación más frecuente es la p.Phe508del (c.1521_1523delCTT, según la nueva nomenclatura estandarizada siguiendo las recomendaciones de la Human Genome Variation Society, www.hgvs.org). Alrededor del 70% de los alelos FQ a nivel mundial contienen esta mutación. Consiste en la pérdida de un codón que lleva a la ausencia de una fenilalanina en la posición 508 de la proteína. Esta proteína alterada no alcanza su ubicación correcta en la membrana celular, quedando retenida en el aparato de Golgi, y es destruida por el proteasoma. Es una mutación de clase II.
Además de la mutación p.Phe508del, en la mayoría de las poblaciones existen otras mutaciones frecuentes, cada una de ellas alcanzando frecuencias de aproximadamente 1-2%, como son p.Gly542* (c.1624G>T), p.Gly551Asp (c.652G>A), p.Arg553* (c.1657C>T), p.Trp1282* (c.3846G>A) y p.Asn1303Lys (c.3909C>G. Existen mutaciones específicas de ciertas poblaciones que en ellas alcanzan frecuencias superiores al 30%. En la mayoría de las poblaciones, todas esas mutaciones combinadas cubren aproximadamente el 75-85% de todos los alelos mutados. El grupo restante de alelos mutados en una población particular comprende mutaciones muy poco frecuentes, algunas de ellas solo se encuentran en una única familia (tabla 3).
Distribución de las mutaciones de CFTR en las poblaciones con mayor prevalencia
| Nombre de la mutación | Exón/intrón | Frecuencia | (%) | Población con la mayor prevalencia |
|---|---|---|---|---|
| p.Phe508del | 11 | 28.948 | 66 | |
| p.Gly542* | 12 | 1.062 | 2,4 | Española |
| p.Gly551Asp | 12 | 717 | 1,6 | Inglesa |
| p.Asn1303Lys | 24 | 589 | 1,3 | Italiana |
| p.Trp1282* | 23 | 536 | 1,2 | Judío-askenazi |
| p.Arg553* | 12 | 322 | 0,7 | Alemana |
| c. 489+1G>T | 4 | 315 | 0,7 | Canadiense-francesa |
| c. 1585-1G>A | 12 | 284 | 0,6 | Italiana |
| p.Arg117His | 4 | 133 | 0,3 | |
| p.Arg1162* | 22 | 125 | 0,3 | Italiana |
| p.Arg347Pro | 8 | 106 | 0,2 | |
| c.3718-2477C>T | i 22 | 104 | 0,2 | |
| p.Ile507del | 11 | 93 | 0,2 | |
| p.Leu88Ilefs*22 | 3 | 78 | 10-30%* | Norte de Finlandia |
| p.Gly85Glu | 3 | 67 | ||
| p.Ala455Glu | 10 | 62 | ||
| p.Phe316Leufs*12 | 8 | 57 | ||
| c.2657+5G>A | i16 | 54 | Española | |
| p.Lys1177Serfs*15 | 22 | 54 | ||
| p.Ard334Trp | 8 | 53 | ||
| c.1766+1G>A | i 13 | 53 | ||
| c.579+1G>T | i 5 | 49 | Canadiense-francesa | |
| p.Lys684Serfs*38 | 14 | 40 | Italiana | |
| p.Leu1258Phefs*7 | 23 | 38 | 6-17%* | Suíza, amish |
| p.Ser549Asn | 12 | 30 | ||
| p.Lys684Asnfs*38 | 14 | 29 | ||
| p.Trp79Leufs*32 | 3 | 87,5%* | Judía-georgiana | |
| p.Met1101Lys | 20 | 69%* | Huteritas | |
| p.Tyr122* | 4 | 48%* | Francesa, Isla Reunión | |
| c.1766+5G>T | i 13 | 30% | China, taiwanesa | |
| c.2988+1G>A | i 18 | 11% | Afroamericana |
Datos obtenidos de CF-Genetic Analysis Consortium. La frecuencia está basada en el screening de 43.849 cromosomas FQ. Las mutaciones se han encontrado en pacientes de origen caucásico, excepto en los así indicado. Para algunas de las mutaciones con mayor prevalencia en una determinada población se indica la localización geográfica (o grupo étnico).
La FQ en los primeros años de su identificación como enfermedad se asoció a un cuadro clínico de malnutrición, diarrea crónica, bronquiectasias, lesiones de fibrosis y quistes pancreáticos, hallados en las necropsias de los pacientes fallecidos.
La afectación pulmonar es la causa más importante de discapacidad grave y muerte prematura en las personas con FQ. Los pulmones de los pacientes son, al nacimiento, histológicamente normales, sin embargo los estudios realizados en muestras de lavados broncoalveolares de lactantes, diagnosticados mediante programas de cribado neonatal, han demostrado que en muchos de ellos, incluso asintomáticos, hay ya evidencias de infección e inflamación.
Manifestaciones clínicas pulmonaresLas manifestaciones pulmonares consisten, en general, en tos y respiración sibilante crónicas asociadas a infecciones pulmonares crónicas o recidivantes. Si el diagnóstico es precoz y se instaura el tratamiento apropiado los pacientes pueden permanecer asintomáticos durante muchos años. No obstante durante la adolescencia o durante los primeros años de la tercera década, la mayoría de los pacientes desarrollan una tos persistente, asociada, generalmente, al comienzo de la infección crónica por P. aeruginosa.
A este inicio de sintomatología clínica persistente se asocian las bronquiectasias y el empeoramiento de la función respiratoria con un patrón obstructivo, que podrá exacerbarse ante infecciones virales con aumento de la tos, aumento de la expectoración habitual, hemoptisis, anorexia y disminución de la capacidad para el ejercicio físico. Con el paso del tiempo la afectación aguda y crónica del parénquima pulmonar produce una lesión del tejido, fibrosis extensa y cambios en la mecánica del pulmón y de las vías respiratorias3. Las lesiones pulmonares evolucionan hacia la insuficiencia respiratoria y hacia el cor pulmonale, que son las causas de muerte más frecuentes en los pacientes con FQ si no reciben un trasplante pulmonar9.
Manifestaciones clínicas gastrointestinalesLa insuficiencia pancreática produce mala absorción de grasa y proteínas y conduce a la malnutrición y a un desarrollo insuficiente de los niños con retardo en la ganancia de peso y estatura. Se suele manifestar en las primeras etapas de la vida y puede ser progresiva.
La afectación hepatobiliar es frecuente en los pacientes adultos y, en estos, es la segunda causa de mortalidad más común, después de la enfermedad pulmonar. Pueden existir colestasis crónica, inflamación, fibrosis e incluso cirrosis.
El íleo meconial aparece en el 10-20% de los recién nacidos como primera manifestación de la enfermedad15. Es casi siempre una manifestación precoz de la fibrosis quística. También puede asociarse a un retraso de la expulsión neonatal de meconio o a un síndrome del tapón meconial16,17. Una gran parte de los recién nacidos con este trastorno de la motilidad intestinal cursan con niveles normales en suero de TIR comportándose como falsos negativos del cribado bioquímico de la FQ.
Manifestaciones clínicas genitourinariasLos varones con FQ suelen ser estériles debido tanto a la ausencia congénita bilateral de los conductos deferentes como a la atrofia de los mismos16, produciéndose azoospermia y disminución del volumen eyaculado.
En las mujeres la deficiencia en la fertilidad está afectada por la presencia de un moco espeso en el tracto genital16. Algunas niñas pueden desarrollar amenorrea e infertilidad. Sin embargo, las mujeres con FQ pueden concebir y tener hijos sanos, siendo los riesgos maternos y fetales dependientes de la gravedad de la enfermedad pulmonar y de sus complicaciones.
DiagnósticoLas recomendaciones actuales según la Cystic Fibrosis Foundation (CFF) para el diagnóstico de FQ son una combinación de sospecha clínica, pruebas bioquímicas y estudio genético18. Los criterios para el diagnóstico de la FQ incluyen la existencia de al menos una características fenotípica, antecedentes familiares o un resultado de cribado neonatal positivo. Por otro lado, es necesario evidenciar la disfunción de CFTR, ya sea por un valor de test de sudor positivo en dos ocasiones o una diferencia de potencial nasal anormal.
Diagnóstico clínicoExamen del cloro en sudor (test del sudor)Este test fue desarrollado en 1959 por Gibson y Cooke6, y sigue siendo una prueba fundamental en el diagnóstico de la FQ. Para realizarlo, se estimula localmente la sudoración con pilocarpina y se determina la concentración de Cl− en el sudor.
Según los criterios clásicos para el diagnóstico de la enfermedad, basados en este test, se considera un valor positivo una concentración de Cl− superior a 60mmol/L. Es infrecuente encontrar concentraciones de Cl− entre 30 y 59mmol/L en niños sin FQ por lo que estos valores deben considerarse «sospechosos» y exigen repetir el test y confirmar o descartar la enfermedad por otros métodos. Por debajo de 30mmol/L de Cl− el test se considera normal.
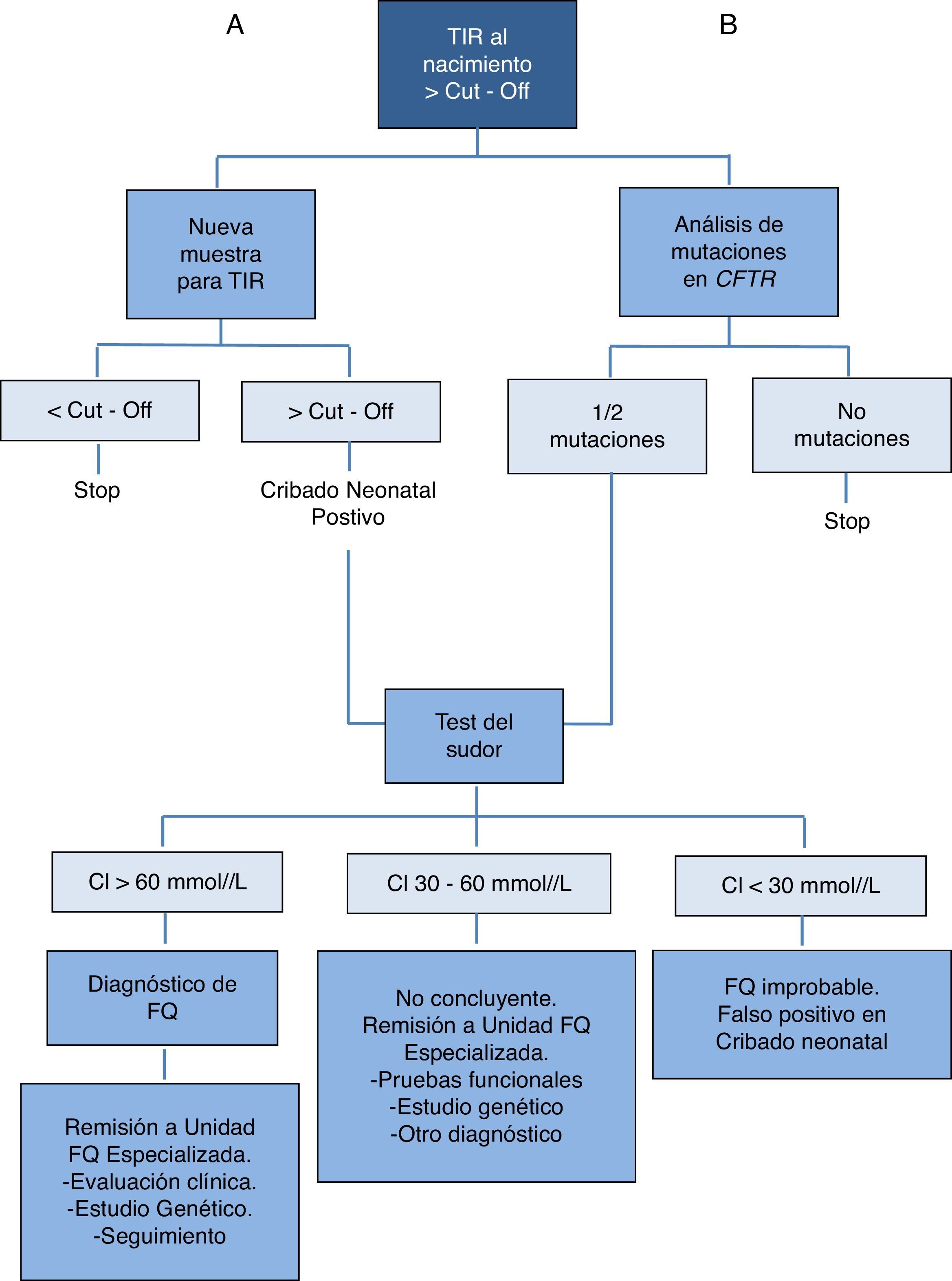
Cribado neonatal de la FQConcentración sérica de tripsina inmunorreactiva (TIR)La medición de la TIR se realiza desde principios de los años setenta19. Los niveles de TIR en suero se elevan en las primeras semanas de vida en los recién nacidos con FQ, hecho que puede ser debido al bloqueo de los conductos pancreáticos exocrinos. Sin embargo, un nivel elevado de TIR al nacimiento no es específico de la FQ, ya puede haber recién nacidos sanos que muestren elevaciones temporales transitorias de esta enzima. Debido a que en la primera semana de vida, cuando se toman la mayoría de las muestras sanguíneas a los recién nacidos, la especificidad de una única TIR elevada es baja, se han desarrollaron protocolos basados en una segunda prueba de TIR con análisis posteriores del DNA y/o test del sudor para reducir los casos falsos negativos y los diagnósticos falsos positivos asociados con la elevación de la TIR20 (fig. 4). En base a estos protocolos, se informaron sensibilidades de entre 85 y 90% (que convencionalmente se calculan con la exclusión de lactantes con íleo meconial).

Uno de los protocolos de actuación en el diagnóstico de la FQ propuestos en la European best practice guidelines for cystic fibrosis neonatal screening. Fuente: Castellani et al.21.
En la actualidad el algoritmo de diagnóstico más aplicado es el TIR-DNA18,21, que consiste en buscar las mutaciones genéticas en el ADN del recién nacido tras el hallazgo de la concentración de TIR elevada. Este método es rápido y no crea ansiedad en las familias, pero su coste es elevado y se detectan muchos «portadores» de una mutación genética. En algunas Comunidades Autónomas se realiza el algoritmo en «dos pasos»: TIR-TIR-DNA. Tras la primera TIR elevada se solicita una segunda muestra que debe extraerse entre los 21 y 30 días de vida, si la TIR de esta segunda muestra es elevada se realiza el estudio de mutaciones genéticas. Esta metodología también tiene algunos inconvenientes, el más importante es la ansiedad provocada a las familias tras la solicitud de una segunda muestra y que tendrán que esperar varios días para conocer el resultado. Este protocolo exige un gran tacto en la información a las familias. En ambos casos debe concluirse con la realización del test del sudor.
Diagnóstico molecularDebido a la existencia de mutaciones frecuentes y específicas de población en el gen CFTR la mayoría de los laboratorios utilizan pruebas dirigidas a la detección de mutaciones específicas que permiten detectarlas de forma simultánea. Las pruebas de este tipo disponibles en el mercado detectan entre el 75-90% de las mutaciones que producen enfermedad y que se observan en alelos de enfermos con FQ en la mayoría de las poblaciones.
La detección completa de mutaciones en las regiones analizadas solo se consigue mediante la secuenciación completa del gen, tarea lenta y costosa. Para la identificación de las mutaciones no detectadas mediante las pruebas dirigidas a la detección de mutaciones específicas se recurre a métodos de rastreo de mutaciones22, como son las técnicas de polimorfismo de conformación de cadena sencilla (Single Stranded Conformation Polymorphism), electroforesis en gel con gradiente de desnaturalización (Denaturing Gradient Gel Electrophoresis), cromatografía líquida de desnaturalización a alta resolución (Denaturing High Performance Liquid Chromatography) y, más recientemente, la fusión de alta resolución (High Resolution Melting). Mediante las técnicas de rastreo de mutaciones se busca secuenciar únicamente las regiones relevantes del gen que muestran un comportamiento anormal en una de estas pruebas y poder así confirmar y caracterizar la identidad de la mutación real.
En los últimos años, la aparición de técnicas de secuenciación masiva (Next Generation Sequencing) ha supuesto un gran avance en el diagnóstico molecular de la enfermad. La Next Generation Sequencing permite obtener millones de secuencias de ADN a una velocidad sin precedentes y a un coste cada vez más reducido, con lo que se prevé que en un futuro pueda desplazar a los métodos moleculares clásicos.
Para el clínico, en ocasiones, puede resultar complicada la interpretación de los resultados de genética molecular y la integración de estos en el proceso diagnóstico. El diagnóstico se basa ante todo en la presentación clínica, y se apoya en la evaluación de la función de CFTR (test del sudor) y el análisis genético. Ninguna de estas características es suficiente por sí misma para hacer un diagnóstico de FQ. Las asociaciones genotipo/fenotipo son útiles en la realización de estudios epidemiológicos, pero el genotipo FQ no puede predecir con exactitud el resultado de forma individual, por lo que su uso en el pronóstico de un enfermo de FQ no está recomendado. La comunicación entre el clínico y el laboratorio de genética es de vital importancia y el informe debe emitirse de forma comprensible tanto para el clínico como para el paciente.
TratamientoNo existe, de momento, una terapia curativa para la FQ. El tratamiento de esta enfermedad está basado en tres pilares fundamentales que son:
- 1.
Conseguir una nutrición adecuada.
- 2.
Utilizar medicamentos que luchen contra la infección e inflamación respiratoria.
- 3.
Realizar con regularidad la terapia física consistente en fisioterapia respiratoria, ejercicios de fortalecimiento de la musculatura del tórax para prevenir deformidades, así como la práctica de algún deporte23.
La fisioterapia respiratoria y el tratamiento antimicrobiano son las dos piedras angulares del tratamiento. Los avances y mejoras en el empleo de los antibióticos son los que han tenido un peso mayor en el aumento de la supervivencia de los pacientes en las últimas décadas.
Ante el primer signo de afectación pulmonar, se recomienda iniciar la fisioterapia respiratoria. En los casos de obstrucción reversible de la vía aérea respiratoria, pueden administrarse broncodilatadores y corticoides en aerosol. La oxigenoterapia está indicada en los pacientes con insuficiencia pulmonar grave e hipoxemia. La ventilación con presión positiva no invasiva con mascarilla facial o nasal también puede resultar beneficiosa en los casos avanzados de insuficiencia respiratoria.
Los antibióticos inhalados mejoran la función pulmonar y disminuyen el tiempo de estancia hospitalaria, en el caso de infecciones por Pseudomonas19.
Los corticoides orales están indicados en los lactantes con bronquiolitis prolongada y en los pacientes con broncoespasmo rebelde, aspergilosis broncopulmonar alérgica y complicaciones inflamatorias (p. ej., artritis, vasculitis).
Tratamiento de la afectación del aparato digestivoEl tratamiento más generalizado es la administración de enzimas pancreáticas (para mejorar la absorción de los alimentos), suplementos alimentarios y complejos vitamínicos de aquellas vitaminas que puedan ser deficitarias.
Se recomienda una buena hidratación permanente por vía oral para evitar la sequedad de las secreciones, especialmente en los períodos de diarrea e infecciones intestinales. Se debe de añadir un suplemento de sal durante los períodos febriles, cuando aumenta la sudoración.
En el caso de que se presente obstrucción intestinal por íleo meconial la obstrucción puede vencerse, en algunas ocasiones, con enemas o puede ser necesaria una intervención quirúrgica.
Terapias futuras en investigaciónTerapia génicaLa terapia génica consiste en la inserción de un gen «funcionalmente normal» en una célula huésped por medio de un vector apropiado. La transferencia del gen CFTR normal a células epiteliales cultivadas utilizando adenovirus como vectores se consiguió a principios de los noventa, pero cuando se ensayó en humanos la respuesta inmunológica del huésped ante la expresión de proteínas virales produjeron efectos adversos inadmisibles24. En la actualidad se investiga con nuevos vectores con resultados prometedores en cuanto a la seguridad, estabilidad y eficacia en su utilización in vivo25.
Terapia con chaperonas reparadoras o estabilizadoras de proteínasLas chaperonas son proteínas presentes en todas las células cuya función es la de unirse a otras proteínas recién formadas y ayudar a su plegamiento, ensamblaje y transporte a la parte de la célula donde estas nuevas proteínas realizan su función. También se investiga con moléculas que ayudarían a la proteína CFTR a mejorar su función como canal de iones Cl-26.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
