



La ataxia con apraxia oculomotora e hipoalbuminemia severa (AOA/EAOH) es una entidad rara que se caracteriza por un inicio temprano de ataxia cerebelosa de progreso lento, seguida por apraxia oculomotora y neuropatía periférica severa que lleva a la cuadriplejia. Pertenece al grupo de ataxias autsómocas recesivas (ARCA), siendo la de mayor frecuencia tras la ataxia de Friedreich, y la primera en población japonesa. El diagnóstico definitivo es mediante el estudio molecular del gen APTX que codifica para la aprataxina.
Se presenta el caso de una mujer de 52 años, con síntomas y datos sugerentes desde la infancia que ha sido diagnosticada 30 años después.
Ataxia with oculomotor apraxia and severe hypoalbuminemia (AOA/EAOH) is a rare entity characterized by early onset of slowly progressive cerebellar ataxia, followed by oculomotor apraxia and peripheral neuropathy leading to severe quadriplegia. AOA/EAOH belongs to the group of autosomal recessive ataxias, being the most frequent after Friedreich's ataxia. Definitive diagnosis provided by identification of mutations in the APTX gene encoding aprataxin.
This case study of a 52 year-old woman with childhood onset of the disease, suggested by symptoms and several additional findings, who has been diagnosed 30 years later.
La hipoalbuminemia es una situación clínica en la cual existe, una disminución del nivel sérico de albúmina por debajo de 3,5g/dL. Algunos autores la clasifican como «severa» cuando es menor de 2,2g/dL. Existen múltiples causas que pueden desencadenar un descenso en la concentración de albúmina plasmática, y que se pueden clasificar en cuatro categorías: disminución de la síntesis, aumento del catabolismo, aumento de las pérdidas y alteración de la distribución1–3.
La hipoalbuminemia por disminución de la síntesis (insuficiencia hepática, desnutrición) o aumento del catabolismo (procesos inflamatorios) tiende a ocurrir en un período relativamente largo de semanas o meses. Dado que la albúmina plasmática tiene una vida media de 17 a 20 días y que solo un 20-30% de los hepatocitos sintetizan albúmina en un determinado momento, existe una gran capacidad de reserva en estas situaciones1–3.
Las pérdidas de albúmina se producen por hemorragias, nefropatías, enteropatía perdedora de proteínas y pérdidas exudativas (quemaduras, dermatopatías y drenajes quirúrgicos). Una disminución significativa de la albúmina sérica como consecuencia de estas pérdidas, puede ocurrir en cuestión de horas1–3.
La alteración en la distribución de la albúmina entre los compartimentos intravascular y extravascular es probablemente la causa más frecuente de la hipoalbuminemia en el paciente crítico. Debido al aumento en la permeabilidad capilar puede producirse una salida rápida de la albúmina al espacio intersticial. El movimiento promedio de la albúmina es solo de 16 horas, por lo que los cambios en la distribución de la albúmina pueden llevar a hipoalbuminemia en pocas horas, como sucede por ejemplo en la sepsis4.
La consecuencia más sensible de la hipoalbuminemia es el desarrollo de edemas, debido a la disminución de la presión oncótica del plasma; y si esta es muy importante pueden aparecer también, ascitis y derrame pleural5.
Esporádicamente aparecen hipoalbuminemias severas, que paradójicamente cursan con edemas parciales e incluso ausencia de edema. Es el caso de la analbuminemia congénita, antes hipoalbuminemia idiopática. Bennhold et al. describieron el primer caso en 19546.
Caso clínicoMujer de 53 años, sin antecedentes familiares de ataxia. Hija única de padres consanguíneos. Ingresa en Medicina Interna en 2007, para estudio de edema intenso en miembros inferiores. Fue diagnosticada de ataxia de Friedreich a los 38 años en 1993.
De la revisión de la historia clínica y los informes aportados por la paciente, observamos que existe ataxia de inicio temprano (antes de los 8 años); neuropatía periférica de predominio distal con déficit motor que condujo a cuadriplejia a los 45 años; marcada atrofia cerebelosa; hipoalbuminemia persistente; pies cavos y escoliosis. No presenta retraso mental.
Al ingreso y tras las exploraciones y estudios realizados, se encuentra: edemas en miembros inferiores, con pulsos periféricos presentes; movilidad abolida con arreflexia generalizada e hipoestesia en las 4 extremidades; severa dismetría bilateral; obesidad y marcada lipodistrofia; no presenta ascitis ni cardiopatía y se observa nistagmus marcado.
En cuanto a la enfermedad degenerativa de la paciente, no puede excluirse el diagnóstico previo de ataxia de Friedreich, pero llama la atención la intensa afectación neuropática y la atrofia cerebral, por lo que debería tomarse en cuenta la posible existencia y/o solapamiento de otras ataxias sensitivo-motoras hereditarias.
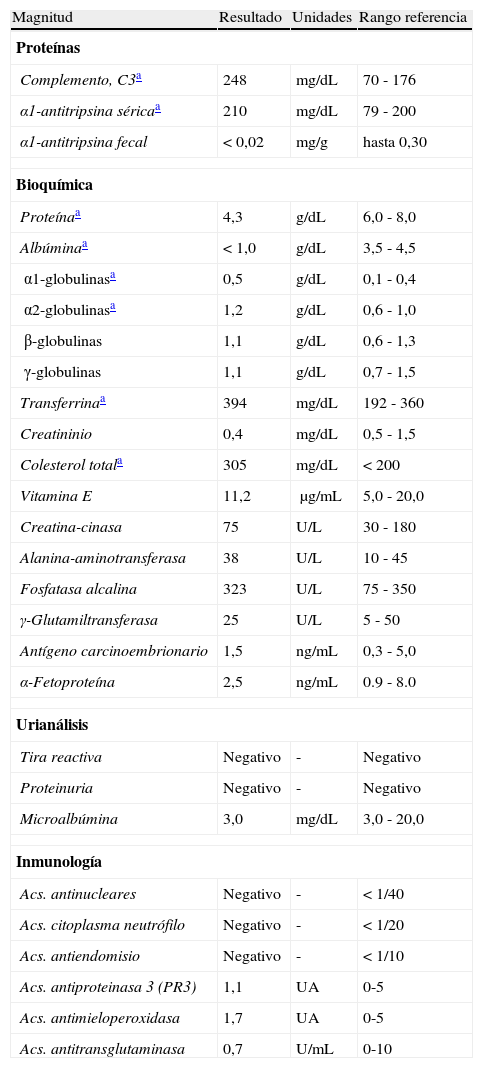
En el estudio de edemas no se encuentran síntomas clínicos ni resultados de laboratorio coherentes con pérdida de proteínas, malnutrición, malabsorción, enfermedad autoinmune, parasitosis, ascitis, neoplasia, proceso inflamatorio agudo, etc. Desde el punto de vista bioquímico, el hallazgo más relevante es la hipoalbuminemia severa (<1g/dL), presente desde hace años (tabla 1). Los estudios de imagen e histopatológicos no revelan nada anormal.
Resultados de laboratorio
| Magnitud | Resultado | Unidades | Rango referencia |
| Proteínas | |||
| Complemento, C3a | 248 | mg/dL | 70 - 176 |
| α1-antitripsina séricaa | 210 | mg/dL | 79 - 200 |
| α1-antitripsina fecal | < 0,02 | mg/g | hasta 0,30 |
| Bioquímica | |||
| Proteínaa | 4,3 | g/dL | 6,0 - 8,0 |
| Albúminaa | < 1,0 | g/dL | 3,5 - 4,5 |
| α1-globulinasa | 0,5 | g/dL | 0,1 - 0,4 |
| α2-globulinasa | 1,2 | g/dL | 0,6 - 1,0 |
| β-globulinas | 1,1 | g/dL | 0,6 - 1,3 |
| γ-globulinas | 1,1 | g/dL | 0,7 - 1,5 |
| Transferrinaa | 394 | mg/dL | 192 - 360 |
| Creatininio | 0,4 | mg/dL | 0,5 - 1,5 |
| Colesterol totala | 305 | mg/dL | < 200 |
| Vitamina E | 11,2 | μg/mL | 5,0 - 20,0 |
| Creatina-cinasa | 75 | U/L | 30 - 180 |
| Alanina-aminotransferasa | 38 | U/L | 10 - 45 |
| Fosfatasa alcalina | 323 | U/L | 75 - 350 |
| γ-Glutamiltransferasa | 25 | U/L | 5 - 50 |
| Antígeno carcinoembrionario | 1,5 | ng/mL | 0,3 - 5,0 |
| α-Fetoproteína | 2,5 | ng/mL | 0.9 - 8.0 |
| Urianálisis | |||
| Tira reactiva | Negativo | - | Negativo |
| Proteinuria | Negativo | - | Negativo |
| Microalbúmina | 3,0 | mg/dL | 3,0 - 20,0 |
| Inmunología | |||
| Acs. antinucleares | Negativo | - | < 1/40 |
| Acs. citoplasma neutrófilo | Negativo | - | < 1/20 |
| Acs. antiendomisio | Negativo | - | < 1/10 |
| Acs. antiproteinasa 3 (PR3) | 1,1 | UA | 0-5 |
| Acs. antimieloperoxidasa | 1,7 | UA | 0-5 |
| Acs. antitransglutaminasa | 0,7 | U/mL | 0-10 |
UA: Unidades arbitrarias.
Al no poder establecerse el origen de la hipoalbuminemia, se realiza una revisión bibliográfica exhaustiva para hipoalbuminemia severa/ataxia y se encuentran 2 posibles causas: Analbuminemia y Ataxia de inicio temprano con apraxia ocular e hipoalbuminemia severa (EAOH/AOA).
La analbuminemia [OMIM#103600], es un trastorno autosómico recesivo, en que los individuos afectados (homocigotos) presentan una virtual ausencia de albúmina circulante, pero paradójicamente cursa con edemas parciales e incluso ausencia de edema7,8.
EAOH/AOA forma parte del grupo de ataxias cerebelosas autosómicas recesivas (ARCA), un conjunto heterogéneo de enfermedades que incluyen la ataxia de Friedreich, la de mayor frecuencia relativa (38%)9–11.
AOA se presenta como 2 entidades diferentes: ataxia con apraxia ocular tipo 1 (AOA1) y tipo 2 (AOA2). Los dos tipos son muy similares, pero están causadas por mutaciones en diferentes genes12,13.
AOA1 [OMIM#208920] fue descrita inicialmente en familias japonesas en 1971. Un fenotipo similar fue descrito en familias portuguesas en 2001, lo que permitió obtener el mapa de AOA1 en el cromosoma 9p13, seguido de la identificación de las mutaciones en el gen APTX14–16.
El gen APTX codifica aprataxina, una proteína, cuya función aún se desconoce, pero que podría tener un papel importante en los mecanismos de reparación del ADN. Consta de 7 exones, y hasta la fecha, se han encontrado 16 mutaciones diferentes. Todas las mutaciones identificadas hasta ahora se localizan en los exones 5, 6 y 717–20.
AOA1 se sospecha en individuos con ataxia cerebelosa de inicio temprano, apraxia oculomotora, neuropatía periférica e hipoalbuminemia severa; todo ello presente en el fenotipo de la paciente, por lo que se propone estudio molecular del gen APTX.
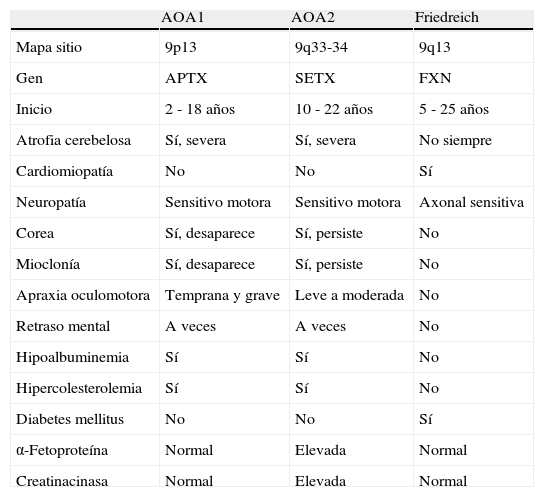
Por otra parte, se cuestiona el diagnóstico de ataxia de Friedreich ya que la paciente no muestra miocardiopatía, pero sí atrofia cerebral severa e hipoalbuminemia21–24. El diagnóstico diferencial incluye asimismo: analbuminemia8, AOA225–28, ataxia con deficiencia de PTT-vitamina E29–31; ataxia tipo SCA2, la cual está asociada a ataxia cerebelosa y movimientos oculares lentos32–34; enfermedad de Charcot-Marie-Tooth 1 con neuropatía sensitivo-motora y pies cavos35,36. Se realizan estudios moleculares específicos con resultados normales. Las diferencias entre AOA1, AOA2 y ataxia de Friedreich se especifican en la tabla 2.
Características diferenciales entre: AOA (AOA1/AOA2) vs ataxia Friedreich
| AOA1 | AOA2 | Friedreich | |
| Mapa sitio | 9p13 | 9q33-34 | 9q13 |
| Gen | APTX | SETX | FXN |
| Inicio | 2 - 18 años | 10 - 22 años | 5 - 25 años |
| Atrofia cerebelosa | Sí, severa | Sí, severa | No siempre |
| Cardiomiopatía | No | No | Sí |
| Neuropatía | Sensitivo motora | Sensitivo motora | Axonal sensitiva |
| Corea | Sí, desaparece | Sí, persiste | No |
| Mioclonía | Sí, desaparece | Sí, persiste | No |
| Apraxia oculomotora | Temprana y grave | Leve a moderada | No |
| Retraso mental | A veces | A veces | No |
| Hipoalbuminemia | Sí | Sí | No |
| Hipercolesterolemia | Sí | Sí | No |
| Diabetes mellitus | No | No | Sí |
| α-Fetoproteína | Normal | Elevada | Normal |
| Creatinacinasa | Normal | Elevada | Normal |
(*) Solo se describen las características genéticas, los aspectos clínicos y los datos de laboratorio que permiten distinguir entre sí las 3 entidades diagnósticas.
Se extrajo el ADN genómico mediante procedimiento automatizado (Chemagic Magnetic Separador Module1), CHEMAGEN®37. El protocolo consiste en la separación de ácidos nucleicos con bolas magnéticas a partir de sangre periférica recogida sobre EDTA como anticoagulante, previo consentimiento informado por parte de la paciente. Se amplificaron mediante reacción en cadena de la polimerasa (PCR) los 7 exones codificantes y las uniones exón-intrón del transcrito con el marco de lectura más largo del gen APTX (número de acceso GenBank NM_175073). Este análisis detecta la práctica totalidad de mutaciones conocidas para APTX. Las condiciones de amplificación consistieron en una desnaturalización inicial de 3 minutos a 95°C, 35 ciclos de 20 segundos a 95°C, 30 segundos a 60°C y 40 segundos a 72°C, con una extensión final de 3 minutos a 72°C. En cada reacción se añadieron 0,75U de GoTaq® DNA Polymerase (Promega), GoTaq® DNA Polymerase buffer 1X, magnesio a 1,5mM, cada cebador a 0,2μM y 50ng de ADN genómico, en un volumen final total de 25 microlitros. Los productos de PCR se purificaron enzimáticamente mediante incubación con ExoSAP-IT® (USB Corporation) 15 minutos a 37°C e inactivación 15 minutos a 80°C.
La secuenciación cíclica bidireccional se realizó con el kit Big Dye® Terminator v3.1 (Applied Biosystems)38, seguida de la purificación de los productos con Montage SEQ96 Sequencing Reaction Cleanup kit (Millipore Corporation)39, y electroforesis capilar con el secuenciador automático 3730XL DNA Analyzer (Applied Biosystems).
Los cebadores utilizados se recogen en la tabla 3. Cada exón se amplificó por PCR con la pareja de cebadores F y R correspondiente (por ejemplo, exón 1: con los cebadores APTX-1F y APTX-1R), posteriormente el producto de PCR se secuenció en el sentido directo con dicho cebador F (forward) y sentido inverso con el cebador R (reverse) correspondiente.
Cebadores utilizados para la amplificación por PCR y para la secuenciación cíclica bidireccional de los siete exones codificantes del gen APTX
| CEBADORES | SECUENCIA (5′->3′) |
| APTX-1F | acttgcctcaagatgaacagc |
| APTX-1R | ccctggtgttggctaaaaga |
| APTX-2F | caataacaggtgattgctaataggc |
| APTX-2R | ccaagcaccttgccttatgt |
| APTX-3F | acataaggcaaggtgcttgg |
| APTX-3R | gattttcacgccacaaacaa |
| APTX-4F | aaggcaatggagtgaagcag |
| APTX-4R | tgccaatgtgaaaagcctct |
| APTX-5F | tcctctctcaaaactgggcta |
| APTX-5R | aggcagaagtgggaactcaa |
| APTX-6F | gcagaggcttttcccatttt |
| APTX-6R | tagctttctaggtcccccaag |
| APTX-7F | atacctggcctggcatactg |
| APTX-7R | ctctacattgtggccactcaa |
Los exones codificantes amplificados han sido numerados de forma consecutiva del 1 al 7. La correspondencia con la denominación exónica de la secuencia referencia de GenBank NM_175073 es la siguiente: exón 1=exón 3, exón 2=exón 4a, exón 3=exón 5b, exón 4=exón 6, exón 5=exón 7, exón 6=exón 8, exón 7=exón 9.
Los electroferogramas se analizaron mediante el paquete informático «Staden Package», diseñado para el ensamblaje, edición y análisis de secuencias de ADN40.
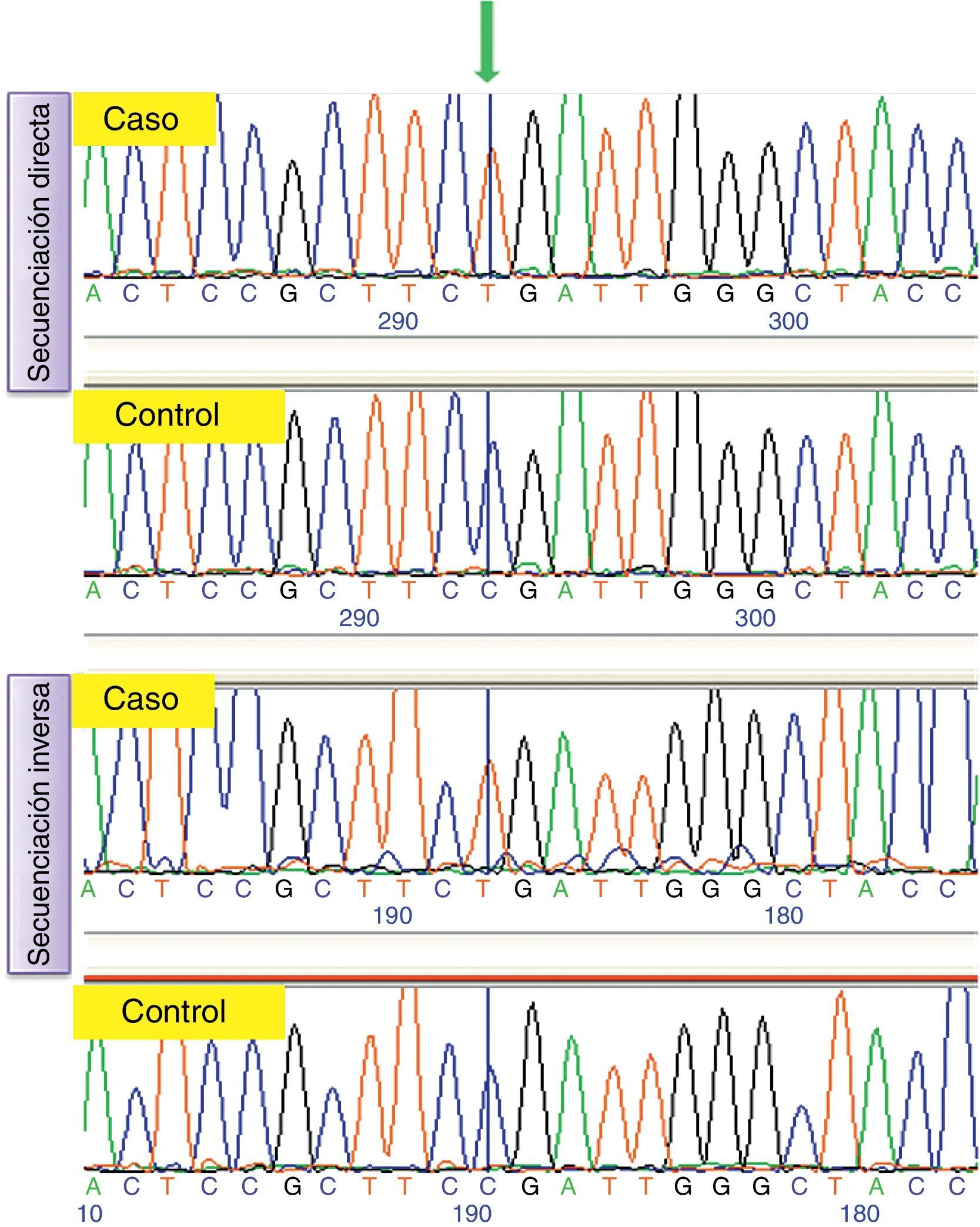
ResultadosLa paciente es portadora de un cambio en homocigosis de una citosina por una timina en el quinto exón del gen APTX (NM_175073.1: c.739C>T), lo que predice la sustitución de un codón codificante para el aminoácido arginina por un codón de parada, produciéndose por tanto una proteína truncada (NP_778243.1: p.R247X). Esta mutación ha sido descrita previamente como causante de AOA1 por Mosesso et al.17.
En la figura 1, se presentan los electroferogramas correspondientes al quinto exón. Se incluye la secuenciación en ambas direcciones del caso a estudio frente a un caso control.
Discusión
La hipoalbuminemia es una situación clínica muy frecuente41. La presencia de edema, hace que en la mayoría de los casos la causa de la hipoalbuminemia pueda ser explicada2,3,5.
Cuando existe hipoalbuminemia marcada sin edema o edema parcial el diagnóstico debe dirigirse hacia analbuminemia congénita o EAOH/AOA. La analbuminemia puede ser fácilmente detectada por los análisis de rutina de laboratorio, como electroforesis del suero42–44. En cuanto a EAOH/AOA, entidad diagnóstica relativamente reciente, el estudio es más complejo, pues síntomas principales como la ataxia y neuropatía requieren diagnóstico diferencial con otras enfermedades y, la apraxia oculomotora puede aparecer tardíamente o pasar inadvertida en exámenes rutinarios o estar solapada con la presencia de nistagmus12,13,45.
En el caso que acabamos de exponer, existió hipoalbuminemia severa durante años, casi siempre sin edemas; pero nunca se investigó su origen por distintas razones: bien porque la situación de hipoalbuminemia sin edemas no es habitual en el contexto clínico; bien porque se considerara normal en la situación de la paciente (inmovilización prolongada, obesidad y lipodistrofia) e incluso porque se pensase en un error de laboratorio.
Es probable que la presencia de síntomas clínicos moderados en relación con la hipoalbuminemia, atribuidos a un efecto compensatorio por el incremento de la síntesis de otras proteínas, que estabilizan la presión oncótica; hizo que no se tomara en cuenta el descenso de albúmina. En este escenario las alteraciones en el metabolismo de los lípidos son muy importantes y consisten en un aumento significativo del colesterol total46,47 (tabla 1).
Los resultados de laboratorio siempre aportan valor añadido al estudio clínico y forman parte importante del proceso diagnóstico, por lo que los informes reiterados por nuestro laboratorio de albumina<1g/dL debieron ser atendidos. La interpretación de la hipoalbuminemia fue sesgada, pues no se tuvo en cuenta cuando no existió edema. Sin embargo, cuando se llevó a cabo el estudio a fondo sobre el origen de la misma se pudo descartar el diagnóstico inicial de ataxia de Friedreich y obtener el diagnóstico definitivo de AOA1, si bien es cierto que todavía no se conoce, a día de hoy la causa por la que se produce un descenso tan significativo de albúmina en AOA1.
La conclusión es que puede presentarse la situación clínica de hipoalbuminemia no explicada por los mecanismos habituales; y que independientemente de la presencia de edema o de su baja incidencia, debería investigarse siempre su origen.
Agradecemos al profesor Lorenzo Minchiotti (Department of Biochemistry «A. Castellani» University of Pavía. Italy) la ayuda que nos ha prestado en el estudio de este caso y la realización del estudio molecular del gen ALB.
