


El término diagnóstico prenatal comprende todas las modalidades de diagnóstico dirigidas a detectar durante la gestación una anomalía congénita que incluya trastornos estructurales o funcionales. Un porcentaje de las mismas se debe a factores genéticos. El presente documento pretende detallar las indicaciones actuales de las pruebas invasivas y de las no invasivas, describir las pruebas de laboratorio que se utilizan en el diagnóstico prenatal de alteraciones genéticas y proponer esquemas de trabajo para el estudio de estas alteraciones genéticas.
The term prenatal diagnosis includes all diagnostic modalities aimed at detecting a congenital anomaly during pregnancy that includes structural or functional disorders. A percentage of them are due to genetic factors. This document intends to detail the current indications of invasive and non-invasive tests, describe the laboratory tests used in the prenatal diagnosis of genetic alterations, and propose work schemes for the study of these genetic alterations.
El término diagnóstico prenatal comprende estrictamente todas las modalidades de diagnóstico dirigidas a detectar durante la gestación una anomalía congénita que incluya trastornos estructurales o funcionales. Aunque la gran mayoría de malformaciones (50-60%) son de causa desconocida, se estima que un 25% puede deberse a factores genéticos1.
Las alteraciones genéticas que se identifican en el diagnóstico prenatal se pueden clasificar en 3grandes grupos2:
- –
Cromosomopatías numéricas o estructurales: alteraciones en el número o estructura de los cromosomas que responden a un desequilibrio genómico, por defecto o por exceso.
- –
Enfermedades monogénicas: causadas por mutación de un solo gen.
- –
Enfermedades poligénicas/multifactoriales: derivadas de la combinación de mutaciones en varios genes y múltiples factores ambientales.
Para el diagnóstico de estas alteraciones genéticas se utilizan tanto técnicas citogenéticas como moleculares. Las técnicas citogenéticas fueron las primeras en emplearse en diagnóstico prenatal y actualmente todavía se realiza el cariotipo para detectar algunas anomalías cromosómicas. Sin embargo, el análisis cromosómico convencional presenta inconvenientes que han promovido el desarrollo de nuevas técnicas que combinan la citogenética con la biología molecular, como la hibridación genómica comparada (CGH)3. Estas nuevas tecnologías han permitido salvar algunas de las limitaciones del cariotipo. Además de estos métodos, se han desarrollado técnicas que no necesitan realizar cultivos celulares para el diagnóstico rápido de las cromosomopatías más frecuentes. Entre ellas, destaca la reacción en cadena de la polimerasa cuantitativa y fluorescente (QF-PCR) y la hibridación in situ fluorescente (FISH) 4.
El estudio de las alteraciones genéticas en el período prenatal se viene realizando con muestras obtenidas mediante una técnica invasiva, bien biopsia corial, que se obtiene entre las semanas 11-14 de gestación, amniocentesis a partir de la semana 15 o cordocentesis a partir de la semana 20. Estas pruebas conllevan un riesgo de pérdida fetal que se estima alrededor del 0,1% en la amniocentesis, del 0,2% en la biopsia corial y del 3-5% en la cordocentesis5, aunque este porcentaje depende de cada centro. Recientemente, se ha introducido en el ámbito prenatal una nueva alternativa no invasiva, conocida como test prenatal no invasivo (o TPNI), que se lleva a cabo con una muestra de sangre de la madre, y que permite la detección del riesgo de las trisomías más frecuentes y de las cromosomopatías X e Y en el ADN fetal que circula en sangre materna6.
Objeto y campo de aplicaciónEl presente documento pretende detallar las indicaciones actuales de las pruebas invasivas y de las no invasivas, describir las pruebas de laboratorio que se utilizan en el diagnóstico prenatal de alteraciones genéticas y proponer esquemas de trabajo para el estudio de estas alteraciones genéticas.
Indicaciones para la realización del test prenatal no invasivoEl TPNI, por su mayor valor predictivo positivo con respecto al cribado combinado del primer trimestre, se aplica de forma contingente como prueba de segundo nivel para los embarazos:
- -
Con riesgo de aneuploidías en el cribado combinado del primer o segundo trimestre (riesgo entre 1:50 y 1:270 para trisomía 21 y riesgo entre 1:50 y 1:150 para trisomías 13 y 18).
- -
Con riesgo de aneuploidías, por antecedentes de nacido, aborto o interrupción voluntaria del embarazo con cromosomopatía6,7.
Es importante señalar que el TPNI solo está validado como método de cribado de estas alteraciones cromosómicas, pero no para ser utilizado como confirmación diagnóstica, por lo que, de momento, cualquier resultado positivo con TPNI debe ser confirmado mediante una prueba invasiva. En las pacientes con índices de riesgo muy elevados (superior a 1:50), se recomienda de momento realizar directamente prueba invasiva, ya que se ha descrito que en esta población de gestantes, es mayor el porcentaje de alteraciones genéticas que no se detectan por TPNI8. Es posible que en un futuro próximo, cuando se publiquen nuevos datos, el punto de corte para realizar procedimiento invasivo se establezca para riesgos mayores de 1:109.
Hay que tener en cuenta que la sensibilidad y especificidad del TPNI para las cromosomopatías X e Y es dependiente de la metodología aplicada y que las trisomías 18 y 13 se presentan con múltiples alteraciones estructurales detectadas mediante ecografía. En el caso concreto de la trisomía 21, se ha observado que el TPNI aplicado para índices de riesgo más bajos (1:300, 1:500 e incluso 1:1.000) mejora la detección de la trisomía por su mayor sensibilidad (99% vs. 85%)10, aunque eleva considerablemente el número de determinaciones, por lo que, dentro del contexto de la sanidad pública, deberán ser estudios coste/beneficio los que lleven a la toma de decisiones. Se deben señalar, al menos, 2situaciones concretas en que el TPNI ayudaría a mejorar la detección del riesgo de trisomía 21. En primer lugar, las gestantes jóvenes en las que el factor edad puede, al calcular el índice de riesgo, «neutralizar» la medida de la traslucencia nucal (TN) o valores de PAPPA o βhCG muy alejados de la media. En segundo lugar, en aquellas gestantes en las que aparecen marcadores ecográficos en el segundo trimestre, que podrían beneficiarse de la aportación de la mayor sensibilidad de TPNI para la detección del síndrome de Down.
Indicaciones para la realización de pruebas invasivasComo ya se ha mencionado, las técnicas invasivas no están exentas de complicaciones y conllevan cierto riesgo de interferir en la evolución de la gestación, por lo que es esencial seleccionar las gestaciones que pueden beneficiarse de su realización11,12.
Las indicaciones son las siguientes.
- 1.
Progenitor(es) portador(es) o afectado(s) de enfermedades hereditarias mendelianas o monogénicas. En nuestro medio se dan con mayor frecuencia el estudio prenatal de fibrosis quística, poliquistosis renal, distrofia miotónica, síndrome de X frágil, neurofibromatosis, distrofia muscular, corea de Huntington y ataxia de Friedreich. En la actualidad, muchos portadores de estas enfermedades se realizan un diagnóstico genético preimplantacional (DGP).
- 2.
Progenitor(es) portador(es) de alteraciones cromosómicas: translocaciones, inversiones, deleciones o duplicaciones, con el fin de descartar segregaciones desequilibradas. De igual manera, muchos portadores de estas alteraciones se someten a DGP.
- 3.
Riesgo ≥ 1:50 en el cribado combinado de primer trimestre o en cribado bioquímico segundo trimestre. Este punto de corte depende de cada comunidad autónoma. En algunas, es indicación de prueba invasiva el riesgo ≥ 1:270 si no está disponible el TPNI13.
- 4.
TPNI con alto riesgo para trisomías 13, 18 o 21.
- 5.
TN ≥ P99 (o ≥ 3,5mm) en la ecografía del primer trimestre. Este aumento del pliegue nucal se considera un marcador ecográfico primario, es decir, que su presencia aislada es indicación para la realización de prueba invasiva.
- 6.
Los marcadores ecográficos son signos o hallazgos ultrasonográficos transitorios e inespecíficos que representan una variación anatómica de la normalidad, y pueden ser primarios y secundarios
- 7.
Hidropesía no inmune (HNI). La biopsia de corion se lleva a cabo cuando el diagnóstico se realiza en edades gestacionales precoces. La amniocentesis será preceptiva para el estudio de infecciones fetales, así como para posibles estudios de ADN para la investigación de determinadas enfermedades genéticas y metabólicas, como, por ejemplo, el síndrome de Noonan.
- 8.
Presencia de marcadores ecográficos secundarios en el sonograma genético. El sonograma genético es la exploración ecográfica que se realiza con el objetivo de detectar marcadores de cromosomopatías y recalcular el riesgo del cribado combinado, de tal forma que a cada marcador ecográfico le corresponde una likelihood ratio (LR), computándose las LR positivas y negativas14. El riesgo de anomalía cromosómica se incrementa según el número de marcadores secundarios visualizados15-17. Los marcadores secundarios que deben considerarse en el sonograma genético del primer trimestre son los siguientes: ductus venoso (flujo patológico: onda A ausente o reversa), hueso nasal (hipoplasia nasal ≤ 2,5mm o ausencia de hueso nasal) y regurgitación tricuspídea (flujo retrógrado al menos la mitad de la sístole, velocidad > 60cm/s). Sin embargo, el uso rutinario del sonograma genético de primer trimestre no se considera en la práctica obstétrica actual, aunque puede ser aplicado de forma individualizada en riesgos intermedio/borderline o cuando la gestante desea profundizar en el estudio de manera conservadora.
- 9.
Riesgo alto de trisomía 21 después de la realización del sonograma genético de segundo trimestre (entre 19 y 21 semanas). La indicación de sonograma genético en el segundo trimestre también debe ser individualizada y nunca llevarlo a cabo de manera rutinaria, sobre todo si se dispone de cribado combinado. Se debe considerar en gestaciones múltiples que acuden después de las 14 semanas, en gestantes que acuden después de las 19 semanas, etc. Hay controversia sobre los marcadores de segundo trimestre a utilizar en el cribado de trisomía 21. Los mejores parecen ser: pliegue nucal > 6mm, hueso nasal ausente/hipoplásico, ventriculomegalia, arteria subclavia derecha aberrante ARSA (por sus siglas en inglés, aberrant right subclavian artery) e intestino hiperecogénico (la ecogenicidad debe ser similar o superior a la del hueso), que se asocia a fibrosis quística y defectos cromosómicos.
- 10.
Presencia de malformaciones fetales: las malformaciones son defectos de la estructura de un órgano, secundarias a una anomalía primaria específica del desarrollo. El riesgo global de anomalías cromosómicas aumenta en relación directa con el número total de malformaciones detectadas. No obstante, la identificación de una malformación fetal aislada es indicación de prueba invasiva.
- 11.
Restricción del crecimiento intrauterino (RCIU/CIR) precoz (inicio antes de las 24 semanas), severo (< P3) y sin alteraciones en la ecografía Doppler compatibles con insuficiencia placentaria. Se asocia a trisomía 18 y a triploidía.
- 12.
Riesgo de infección fetal por toxoplasma, citomegalovirus, varicela, rubéola, herpes, parvovirus B19 o virus del Zika.
- 13.
Confirmación de un DGP. Existe una probabilidad de error < 5% si el diagnóstico se realizó por FISH y < 1% si fue por PCR, microarray o secuenciación masiva (NGS)
El cariotipo consiste en el estudio cromosómico de células en metafase, momento en el cual los cromosomas se encuentran más condensados.
Las anomalías cromosómicas que se pueden detectar en el período neonatal pueden ser numéricas o estructurales y pueden afectar a uno o más autosomas, a los cromosomas sexuales o a ambos simultáneamente. Las anomalías cromosómicas más frecuentes con repercusión clínica son las anomalías numéricas o aneuploidías, en las que se observa un número de cromosomas anormal debido a un cromosoma extra o ausente. En cuanto a las estructurales, se pueden identificar translocaciones, inversiones y también deleciones o duplicaciones de un segmento cromosómico. Es importante resaltar que, incluso con el aumento de resolución que se ha conseguido en las últimas décadas, no se pueden detectar con el cariotipo deleciones o duplicaciones de tamaño inferior a 4-5 megabases (Mb)18,19.
El cariotipo fetal se lleva a cabo mediante el análisis de células in vitro obtenidas con una prueba invasiva, pudiendo realizarse mediante análisis directo o con células procedentes de cultivo. En la actualidad, la biopsia corial y la amniocentesis son las técnicas invasivas más utilizadas para obtener el material necesario para realizar este cariotipo fetal. Sin embargo, hay que tener en cuenta una posible contaminación de la muestra con células de origen materno tanto por líquidos amnióticos hemáticos como en muestras de vellosidades coriales contaminadas con decidua materna.
Las muestras de líquido amniótico no contienen células fetales en división y tienen que cultivarse in vitro, mediante un cultivo largo para poder obtenerlas en metafase. En el caso de células de vellosidad corial, sí que existen algunas que se dividen espontáneamente, con lo que se pueden observar células en metafase sin necesidad de cultivo, y por tanto, podemos hablar de la realización de un cultivo corto en el cual las metafases obtenidas serán de baja calidad, y de un cultivo largo, de mejor calidad. La calidad y el tamaño de la muestra de vellosidad corial va a variar mucho dependiendo de la posición de la placenta, siendo muy importante evitar tomar la muestra de aquellas regiones contaminadas con decidua materna y vellosidades degeneradas. En el laboratorio, para evitar la contaminación materna es muy importante limpiar las vellosidades antes del cultivo. Tanto en el caso de la vellosidad corial como del líquido amniótico, el factor limitante será el tiempo de cultivo celular, siendo en el caso de cultivo largo un mínimo de 8-10 días y en el caso del cultivo corto 24-48 h, antes de procesar las células para su análisis20.
El análisis del cariotipo estándar implica el estudio del número y estructura de los 46 cromosomas. Se deben contar 20 células en metafase procedentes de 2cultivos independientes y cariotipificar 2células como mínimo. Las anomalías cromosómicas más frecuentes son las que aumentan con la edad materna y se deben a errores de no disyunción, dando como resultado la ganancia o pérdida de un cromosoma. Estas alteraciones pueden observarse tanto en el total de las células como en mosaico (un mosaico se describe como la existencia de 2o más líneas celulares en un mismo individuo)18,19.
Reacción de la polimerasa cuantitativa y fluorescente (QFPCR) e hibridación in situ fluorescente (FISH)Los 2métodos moleculares más empleados en muestras invasivas para el diagnóstico prenatal rápido de las enfermedades cromosómicas frecuentes son la FISH y la QF-PCR. Ambas técnicas usan marcaje con fluorocromos, aunque el abordaje diagnóstico es diferente. Es importante reseñar que normalmente se diseñan para detectar alteraciones genéticas solo en los cromosomas 13, 18, 21, X e Y, que son los más frecuentemente implicados en las aneuploidías, y que permiten ofrecer un resultado en 24-48 h. Se pueden realizar tanto en líquido amniótico como en vellosidad corial.
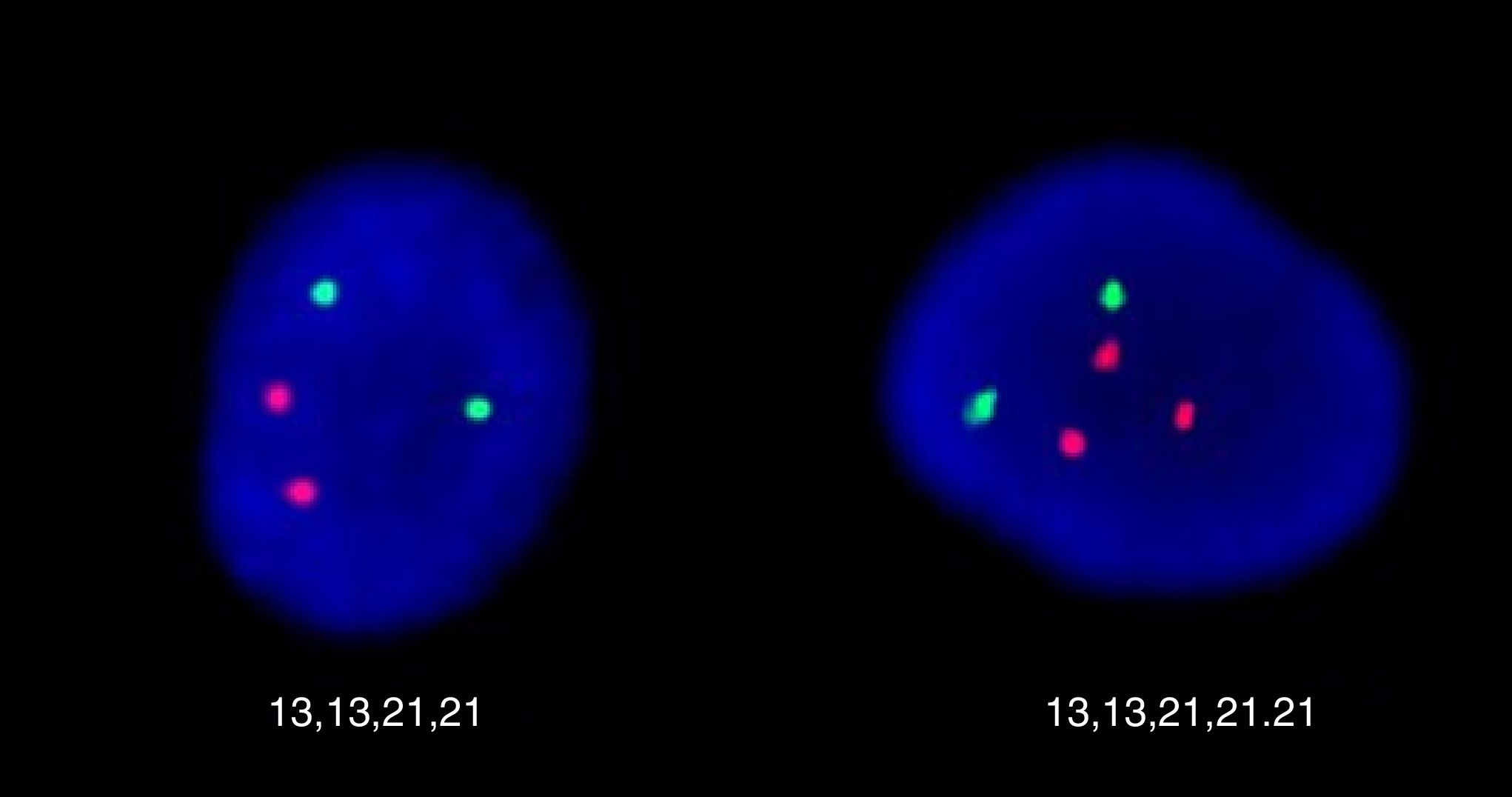
FISHConsiste en la hibridación de secuencias de ADN cromosómicas específicas marcadas con fluorocromos en una preparación cromosómica. Dichas sondas hibridan con su ADN complementario y pueden ser visualizadas al microscopio (fig. 1). La hibridación puede realizarse tanto en células en interfase como en células en metafase.
, color verde y LSI 21 (21q22.13), color rojo, Cytocell. A la izquierda de la imagen observamos un núcleo con 2 señales verdes para el cromosoma 13 y 2 rojas para el 21. El núcleo de la derecha presenta 2 señales para el cromosoma 13 y 3 para el cromosoma 21, hallazgo clínicamente compatible con trisomía 21.")
Hibridación in situ se realiza en células de líquido amniótico sin cultivar, utilizando las sondas de copia única LSI 13 (13q14.2), color verde y LSI 21 (21q22.13), color rojo, Cytocell. A la izquierda de la imagen observamos un núcleo con 2 señales verdes para el cromosoma 13 y 2 rojas para el 21. El núcleo de la derecha presenta 2 señales para el cromosoma 13 y 3 para el cromosoma 21, hallazgo clínicamente compatible con trisomía 21.
La FISH prenatal para el diagnóstico rápido de las aneuploidías más frecuentes se realiza en células sin cultivar, tanto en células de vellosidad corial como de líquido amniótico, utilizándose sondas específicas correspondientes a los cromosomas 13, 18, 21, X e Y.
En el caso de encontrar alguna anomalía en el cariotipo también se puede realizar una FISH con sondas específicas del cromosoma de interés con el objeto de caracterizar dicha anomalía.
El problema que presenta esta técnica es que los cromosomas en interfase están menos condensados que en metafase y dan lugar a señales difusas, lo que en ocasiones dificulta su interpretación. La FISH, con sondas de pequeño tamaño, se observa como 2señales por núcleo para cada cromosoma, en casos diploides, y 3señales en caso de núcleos trisómicos.
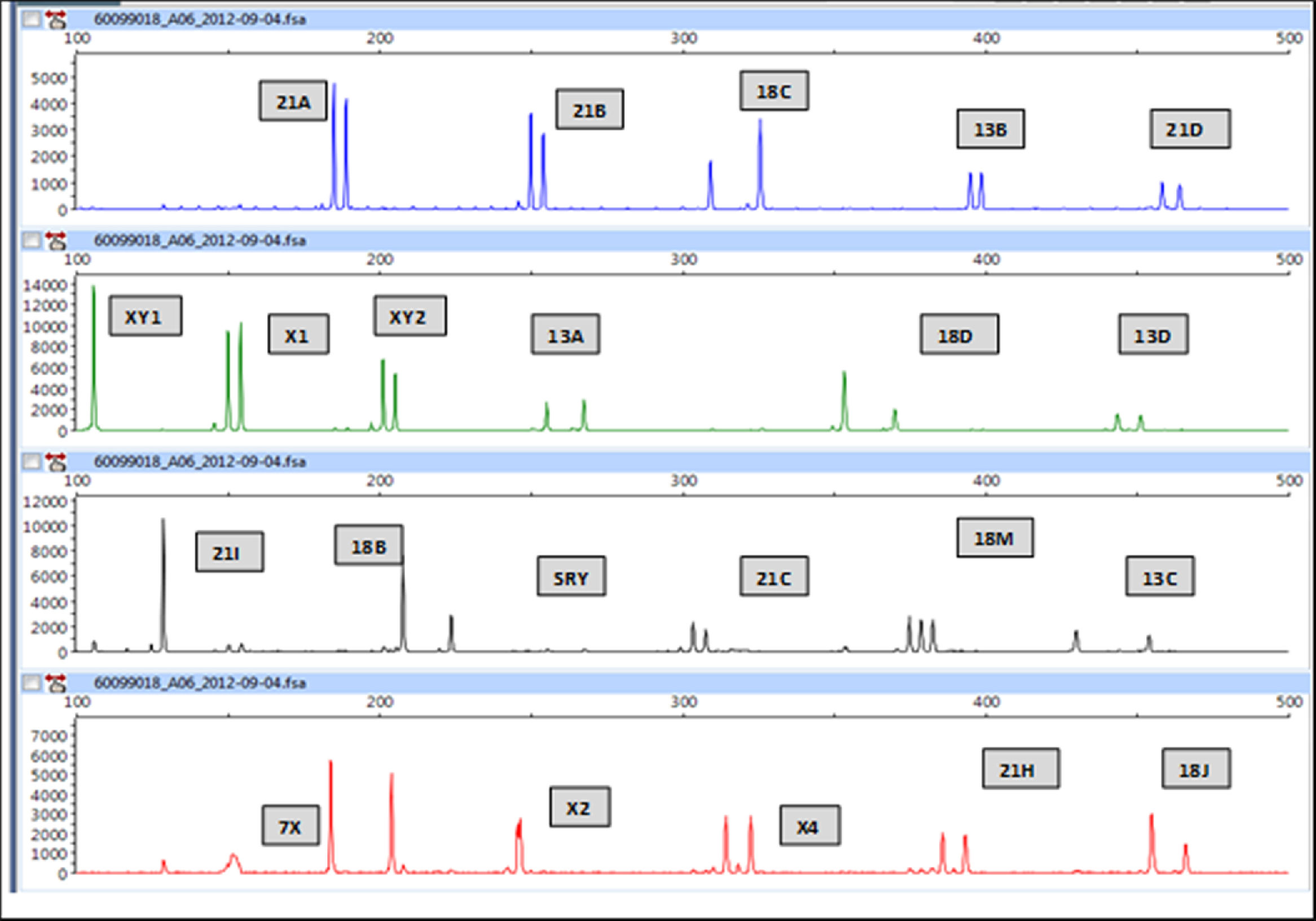
QF-PCRLa QF-PCR es una técnica citogenético-molecular que analiza regiones altamente polimórficas de los cromosomas, llamadas short tandem repeats (STR).
Consiste en la realización de una PCR múltiple utilizando oligonucleótidos marcados con fluorocromos específicos de STR localizados en los cromosomas 21, 18, 13, X e Y. Posteriormente, los productos de PCR se separan y cuantifican mediante electroforesis capilar. Finalmente, los resultados obtenidos se analizan mediante un software específico que calcula la relación del área de los picos obtenidos entre cada alelo, considerándose valores entre 0,8 y 1,4 como normales, al menos para 3marcadores. Marcadores con ratios entre 1,4 y 1,8 o un único pico se consideran no informativos. La presencia de 3alelos (ratio 1:1:1) o 2alelos con una ratio ≤ 0,6 o ≥1,8 (ratio 1:2 o 2:1) se considera como una trisomía para ese marcador (fig. 2). Al menos debe haber 2marcadores informativos con un genotipo anormal para interpretar resultados de un cromosoma en particular como anormales.
 y un patrón trisómico dialélico (1:2 o 2:1) para los marcadores 18C (D18S535), 18B (D18S978), 18D (D18S386) y 18J (D18S535).")
Frente a la técnica de FISH, la QF-PCR presenta varias ventajas21:
- –
La FISH requiere mayor volumen de muestra y mayor tiempo de análisis que la QF-PCR. Para el análisis mediante FISH es necesario analizar un mínimo de 50 núcleos en interfase para cada cromosoma.
- –
Mediante la FISH no es posible determinar la presencia de contaminación materna, puesto que no distingue entre células maternas y células fetales cuando estas son de sexo femenino. Sin embargo, mediante QF-PCR se detectan picos extras o ratios sesgadas entre alelos. Además, existe la posibilidad de analizar los STR de muestra materna y, de ese modo, diferenciar entre los picos.
Por su parte, la FISH tiene la ventaja de permitir descartar/confirmar mosaicos analizando un elevado número de células (un mínimo de 100), mientras que la QF-PCR no detecta mosaicos por debajo del 30%.
Secuenciación SangerLa secuenciación Sanger es ampliamente utilizada en el diagnóstico genético de enfermedades con etiología molecular conocida. Se trata de un método enzimático que va a permitir secuenciar un fragmento de ADN previamente amplificado mediante PCR. La reacción de secuenciación añade dideoxinucleótidos (que sirven como terminadores de la reacción) marcados con fluoróforos mediante la Taq polimerasa y se resuelve mediante una electroforesis capilar. La combinación de estas técnicas es altamente sensible y específica.
Es posible realizar el estudio a partir de biopsia de vellosidades coriales o líquido amniótico. El elevado coste económico y tiempo necesario para realizar el estudio de grandes genes o enfermedades con heterogeneidad genética deben de ser tenidos en cuenta en la secuenciación Sanger. El diagnóstico prenatal requiere un resultado con un tiempo de respuesta de días o semanas, por lo que su aplicación va a estar limitada a determinadas situaciones. La secuenciación Sanger va a estar indicada únicamente en casos con un fenotipo muy específico que sugiera etiología en un único gen y, principalmente, en el estudio prenatal de mutaciones conocidas en padre/s portador/es.
Microarrays cromosómicosLa tecnología de microarrays cromosómicos (CMA) mediante arrays o matrices de CGH permite detectar cambios en el número de copias (deleciones y duplicaciones) o CNV, de las siglas copy number variations, a lo largo de todo el genoma con alta resolución y rapidez.
En cuanto al tipo de muestra, es posible realizar CMA tanto en biopsia de vellosidades coriales, líquido amniótico, sangre u otro tejido fetal. La muestra más habitual suele ser líquido amniótico, ya que se realiza a partir de semana 15 (en las ecografías del segundo trimestre se pueden hacer más evidentes los hallazgos ecográficos) y además presenta menor tasa de mosaicismos confinados a placenta. En paralelo a la extracción de ADN se recomienda realizar un cultivo celular para extraer más ADN o realizar cariotipo u otras técnicas en caso necesario.
Según la tecnología empleada, pueden ser tanto array-CGH (aCGH) como array-single nucleotide polymorphisms (SNP).
- –
En aCGH se produce una hibridación competitiva equimolar de ADN de un paciente (marcado con un fluoróforo, habitualmente Cy5, color rojo) y ADN control del mismo sexo (marcado con otro fluoróforo, habitualmente Cy3, color verde) sobre un soporte físico (cristal), en el que radican moldes de ADN (sondas) con secuencia y localización en el genoma conocida. Las sondas utilizadas como ADN molde pueden ser fragmentos de secuencias extraídas de cromosomas artificiales bacterianos (BAC) o de oligonucleótidos.
- –
En los array-SNP se produce una hibridación no competitiva, comparando la intensidad de hibridación del ADN del paciente con una señal control previamente determinada, en una matriz de SNP. Permite detectar además de cambios en el número de copias, estados de pérdida de heterocigosidad (loss of heterocigosity [LOH]).
Según el diseño, los CMA puede ser dirigidos (sondas localizadas en regiones de interés para enfermedades concretas), de genoma completo (alta densidad de sondas a lo largo de todo el genoma, utilizados sobre todo a nivel de investigación) o de diseño mixto (alta densidad de sondas en regiones de interés y sondas con un nivel de densidad menor en el resto del genoma).
Otro aspecto importante es la resolución del CMA, que es el tamaño mínimo (teórico y real) de una alteración para ser detectada, identificada y mapeada con la mayor precisión posible. Dependerá del tipo del array y del diseño, pudiendo ser distinta en diversas zonas del genoma según la localización de las sondas17,22,23. Algunos autores proponen una resolución mínima global de 400kb, que sería suficiente para detectar variantes patogénicas que motivaron el estudio inicialmente y reducir significativamente el número de variantes de significado incierto encontradas.
El CMA presenta las siguientes limitaciones: no detecta mutaciones puntuales, expansiones de tripletes, reordenamientos equilibrados ni mosaicismos de bajo grado o compensados. Las triploidías y la presencia de contaminación celular materna son detectables por array-SNP, pero no por aCGH. En caso de utilizar aCGH se recomienda realización de QF-PCR16,17.
Es imprescindible detallar los criterios utilizados para la interpretación de CNV, así como el tipo de variantes a informar. Se siguen los criterios del Colegio Americano de Genética Médica y Genómica (ACGM) de clasificación de variantes. Según estos criterios las variantes se clasifican en: patogénica, probablemente patogénica, variante de significado incierto (VOUS), probablemente benigna y benigna24. Hay una tendencia a incluir en el informe solo CNV patogénicas y probablemente patogénicas y solo informar aquellas VOUS con criterios de probable patogenicidad: tamaño, contenido génico y compatibilidad fenotípica16. Cabe hacer mención especial a CNV en loci de neurosusceptibilidad. Se trata de factores de riesgo genético con penetrancia reducida o expresión variable, asociados a discapacidad intelectual, trastornos del espectro autista y alteraciones psiquiátricas. El fenotipo de estas CNV es impredecible, por lo que solo deben informarse en caso de concordancia entre el fenotipo ecográfico y el hallazgo encontrado16,22,25.
Estaría indicada la realización de CMA en diagnóstico prenatal en los siguientes casos:
- 1.
Hallazgos ecográficos:
- –
TN superior percentil 99. Está descrito que un 5-10% de fetos con TN aumentada y cariotipo normal presentan una alteración submicroscópica en el CMA16,17,26-28.
- –
Hallazgo ecográfico asociado a un síndrome concreto. Es necesario evaluar previamente en cada caso la técnica diagnóstica adecuada. En caso de síndromes de microdeleción o microduplicación, el CMA puede ser la técnica de elección17.
- –
Hallazgo ecográfico inespecífico. En caso de malformación ecográfica mayor o retraso de crecimiento intrauterino severo (< percentil 3) y precoz (< 24 semanas), se recomienda CMA como análisis de primera línea, si la QF-PCR es negativa. Entre un 6-9% de estos casos presentan alteraciones en CMA relacionadas con el fenotipo29.
- 2.
Antecedentes personales o familiares:
- –
Presencia de deleción o duplicación familiar críptica. La práctica de CMA es útil en caso de microdeleción o microduplicación con riesgo de transmisión y penetrancia y relevancia clínica significativas16,22,25.
- –
Antecedente familiar de reordenamiento cromosómico en equilibrio. En casos de translocación parental recíproca o inversión pericéntrica, se puede realizar CMA con el fin de descartar segregaciones desequilibradas en los casos en los que no sea detectable por cariotipo16.
- 3.
Presencia en cariotipo fetal de un reordenamiento aparentemente equilibrado de novo o un cromosoma marcador16,23.
La NGS (por sus siglas en inglés, next generation sequencing) no se realiza actualmente en diagnóstico prenatal en la rutina clínica. No obstante, puede estar indicada en determinadas circunstancias. Las aproximaciones diagnósticas normalmente utilizadas en estos casos son el estudio de paneles con genes específicos de una enfermedad (secuenciación dirigida) o la secuenciación del exoma, es decir la secuenciación de las regiones codificantes del genoma. Sin embargo, estos estudios de exoma no se recomiendan fuera de un contexto de ensayos clínicos hasta que se publiquen datos y estudios de validación30, por lo que no hay recomendaciones específicas de su uso en diagnóstico prenatal.
La NGS está indicada en el diagnóstico molecular de enfermedades hereditarias complejas con un fenotipo clínico heterogéneo y con diversidad de genes implicados. Es capaz de detectar mutaciones puntuales, pequeñas deleciones e inserciones y, mediante análisis especiales, permite detectar ganancias y pérdidas de material cromosómico ≥ 7 Mb y pérdidas asociadas a deleciones específicas < 7 Mb, o CNV31.
La secuenciación de exoma puede estar contemplada, por ejemplo, en fetos con múltiples anomalías, o en casos de fenotipos fetales recurrentes en los que no se ha alcanzado un diagnóstico mediante test genéticos convencionales, como son cariotipo o microarray. El ACGM recomienda valorar la secuenciación de exoma cuando no se ha conseguido el diagnóstico con test genéticos específicos para un fenotipo, incluyendo test de secuenciación dirigidos (entre los que se encuentran los paneles de genes de NGS) en un feto con múltiples anomalías congénitas que sugiera origen genético32. De cualquier manera, los datos publicados en diagnóstico prenatal mediante secuenciación del exoma se limitan a pequeñas series de casos y apuntan a que se identifica una anomalía genética hasta en un 20-30% de los fetos con múltiples anormalidades y con resultados normales en los estudios genéticos convencionales33.
La NGS presenta importantes limitaciones. La detección de variantes de significado incierto puede crear ansiedad en la familia y dificultar la toma de decisiones. El conocimiento de las variantes genéticas y sus efectos fenotípicos es todavía limitado y frecuentemente es difícil interpretar las variantes identificadas y no siempre es posible establecer una correlación fenotipo-genotipo.
Esta dificultad en la interpretación de las variantes detectadas, especialmente importante en los estudios de exoma, puede afrontarse en cierta manera con la secuenciación en tríos, que consiste en secuenciar la muestra del feto y de los padres biológicos de manera simultánea.
La posible y frecuente cobertura incompleta en genes claves, la detección de hallazgos incidentales no relacionados con el fenotipo y la posible detección de no paternidades deben ser contempladas en el asesoramiento genético preprueba.
Otras limitaciones, ya de tipo puramente técnico, que existen en la actualidad en la aplicación de la NGS en el diagnóstico prenatal son:
- –
No es capaz de detectar grandes reordenamientos genéticos, variantes en promotores o en regiones intrónicas no incorporadas en la captura. Actualmente, mediante análisis informáticos especiales se están empezando a detectar determinadas mutaciones en mosaico y CNV.
- –
Debido a la baja cobertura obtenida en la NGS en determinadas muestras, nos podemos encontrar con falsos negativos.
- –
El coste es todavía elevado, aunque se está reduciendo considerablemente.
- –
El análisis es laborioso y el tiempo de respuesta generalmente elevado para un diagnóstico prenatal.
Sin embargo, la posibilidad de analizar simultáneamente varios genes y el poder procesar en el mismo ensayo varias muestras reduce considerablemente el coste y el tiempo de procesamiento, haciendo que resulte coste/tiempo eficiente.
Los retos técnicos, éticos y de interpretación que plantea la secuenciación genómica en su aplicación clínica son si cabe mayores en la muestra prenatal, la historia clínica del feto es muy corta y solo la imagen ecográfica contribuye al examen físico del feto. Las características fenotípicas de muchas enfermedades pueden no estar presentes en este período y en el caso de enfermedades letales, nunca se conocerá el fenotipo completo.
Secuenciación masiva en el diagnóstico prenatal no invasivo.El análisis prenatal no invasivo se realiza con los fragmentos de ADN fetal que durante el embarazo circulan en el plasma materno. Aunque ya la PCR a tiempo real y, de forma más específica y sensible, la PCR digital, de mayor sensibilidad que la PCR a tiempo real, habían permitido la detección de secuencias del feto no presentes en la madre (secuencias del cromosoma Y en el feto varón, alelo paterno mutado en un feto portador o afecto, factor Rh fetal en una madre negativa), solo la enorme sensibilidad y especificidad, capacidad de «recuento» de las regiones cromosómicas y poder de procesamiento de datos de la NGS ha permitido la validación analítica y clínica de algunas de sus potenciales aplicaciones asistenciales. La detección de las aneuploidías más frecuentes (21, 18, 13 y cromosomas sexuales) en semana 9-10 mediante NGS, apoyada en poderosos algoritmos y requiriendo una mínima fracción fetal (el ADN fetal es inseparable y muy minoritario frente al ADN materno circulante) es ya aplicable al TPNI6,10. Para ello, la especificidad se dirige a regiones cromosómicas estables que permiten evaluar el número de copias, utilizando cebadores que generan una pequeña secuencia (massive parallel genomic sequencing) o sondas específicas para variantes puntuales SNP en los cromosomas analizados (arrays).
Como ya se ha comentado, esta técnica se limita por el momento al ámbito del cribado y requiere confirmación mediante técnica invasiva.
A nivel diagnóstico, la NGS del ADN fetal circulante se puede aplicar para enfermedades monogénicas34,35, en este caso dirigido a las regiones específicas que contienen alteraciones puntuales ya conocidas y con la obvia precaución de que para la discriminación del alelo materno fetal debe recurrirse también a cuantificar el desbalance. Técnicamente, las pequeñas deleciones y regiones repetidas (CNV) plantean problemas para su detección, actualmente CNV < de 5Mb no son detectadas con fiabilidad. En los loci complejos y seudogenes también debe garantizarse una adecuada validación. Actualmente, en el ámbito asistencial, solo es aplicable para alteraciones validadas clínicamente en su implicación causal de la entidad o en un contexto familiar en que el caso índice define los marcadores y alteraciones a seguir.
El informe de resultados de un análisis de TPNI debe incluir siempre los datos de tasa de detección, especificidad y valores predictivos positivo y negativo propios de la técnica aplicada en el laboratorio. Debe evitarse en lo posible una extensión del análisis (otros cromosomas o deleciones/duplicaciones de menor tamaño) que pueda incrementar los falsos positivos, ya que produciría un aumento de los procedimientos invasivos que se pretendían evitar con la implementación del método. Se debe cuantificar la fracción fetal y reflejar en el informe la cuantía de esta fracción. Para reducir al máximo las muestras que quedan sin resultado debe garantizarse una fracción fetal adecuada (generalmente > 3,5-4%) controlando los factores preanalíticos (edad gestacional, procesamiento de la muestra) y la calidad técnica. No siempre debe plantearse una nueva determinación en las muestras sin resultado. Se debe ofrecer directamente una prueba invasiva si la gestante es obesa o si la determinación de TPNI realizada en semana adecuada9,10 no ha obtenido resultado, ya que, en este caso, el riesgo de aneuploidías es superior36.
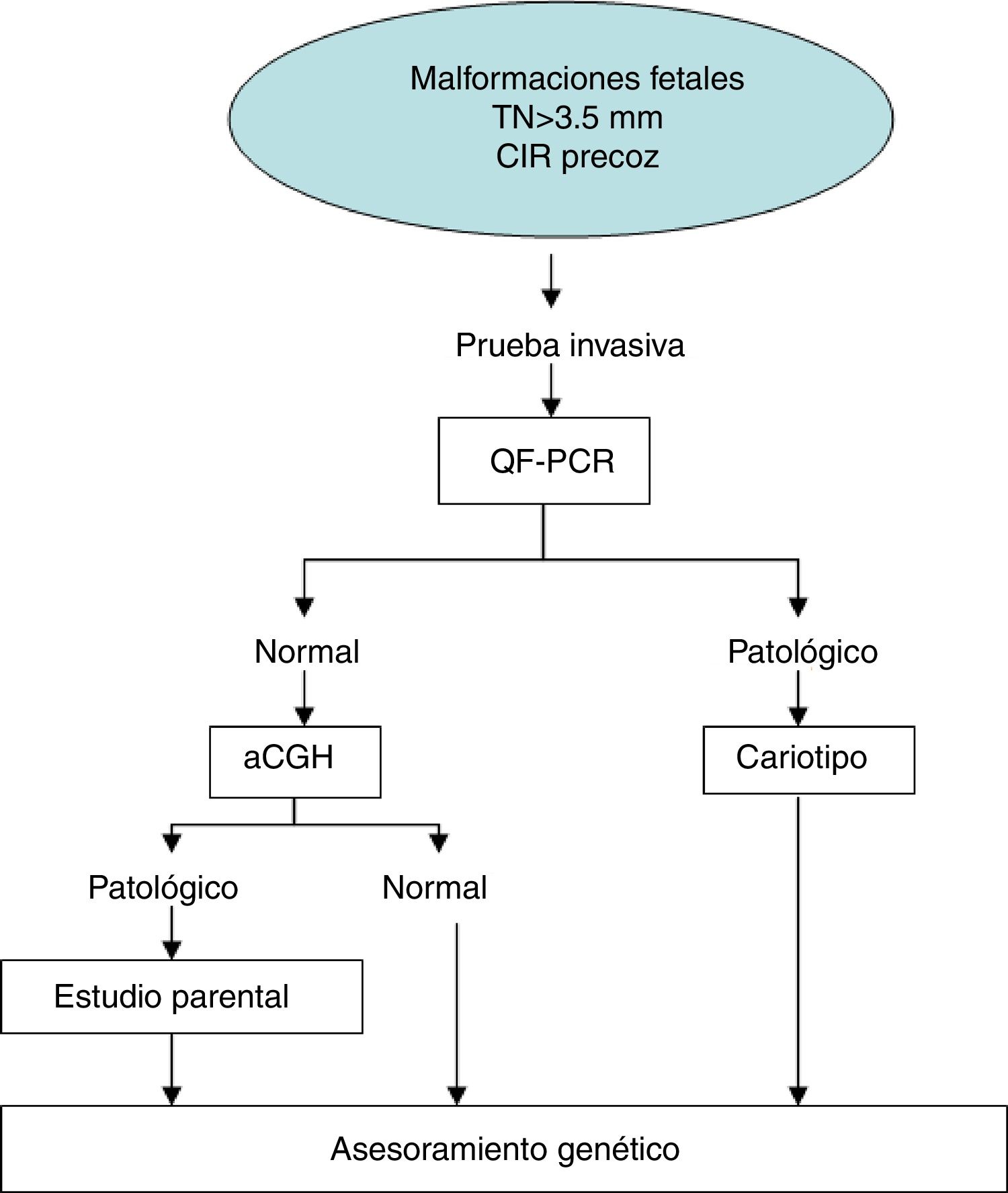
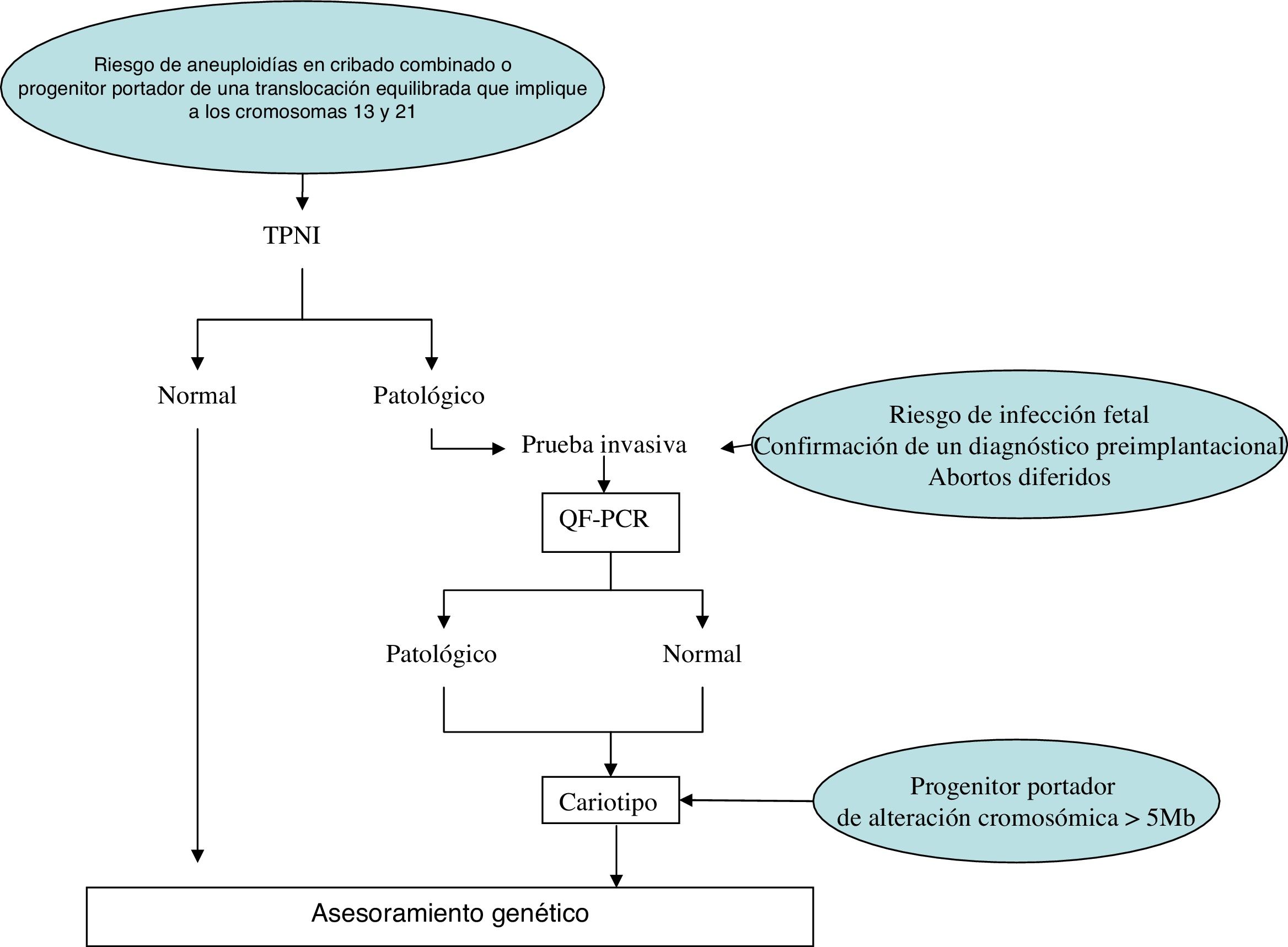
Algoritmo diagnósticoEn las figuras 3-5 proponemos diferentes algoritmos diagnósticos en función de la indicación del estudio genético.


Recomendaciones cuando exista riesgo de aneuploidías en el cribado, haya antecedentes de aneuploidía o riesgo de infección fetal, en abortos diferidos, cuando haya que confirmar un DGP, o cuando algún progenitor sea portador de una translocación equilibrada entre los cromosomas 13 y 21 o portador de cualquier alteración cromosómica equilibrada mayor de 5 Mb.

La QF-PCR se considera actualmente como prueba de primera línea para el estudio prenatal de alteraciones cromosómicas en fetos con alteraciones ecográficas, debido a que las aneuploidías, especialmente la trisomía 21, es la causa más frecuente de anomalía fetal37.
En aproximadamente un 6% de los embarazos que se someten a una prueba invasiva, se identifica una alteración submicroscópica por la técnica de microarray que no puede ser detectada por QF-PCR o cariotipo convencional38. Las guías internacionales recomiendan realizar el estudio de microarray solo en aquellos casos con QF-PCR normal y presencia de anomalía estructural en la ecografía, TN por encima del percentil 99 o CIR precoz25,39,40 (fig. 3). En aquellas gestantes que se sometan a una prueba invasiva por otra razón (confirmación de un DGP, riesgo de infección, etc.) se debe ofrecer, además del estudio que motivó la realización de la prueba invasiva, la QF-PCR y el cariotipo, aunque el riesgo de identificar una alteración genética visible al microscopio sea bajo (fig. 4)25.
El estudio del cariotipo debe también ofrecerse a aquellas pacientes con QF-PCR positiva, para confirmar los resultados y descartar que la presencia de la aneuploidía observada se deba a la existencia de una alteración estructural equilibrada en alguno de los progenitores.
En el caso de que algún progenitor sea portador de una translocación recíproca equilibrada en la que el material intercambiado tenga un tamaño inferior a 5Mb, el estudio prenatal para descartar que el feto haya heredado un derivado desequilibrado de dicha translocación debe realizarse mediante FISH o CGH array.
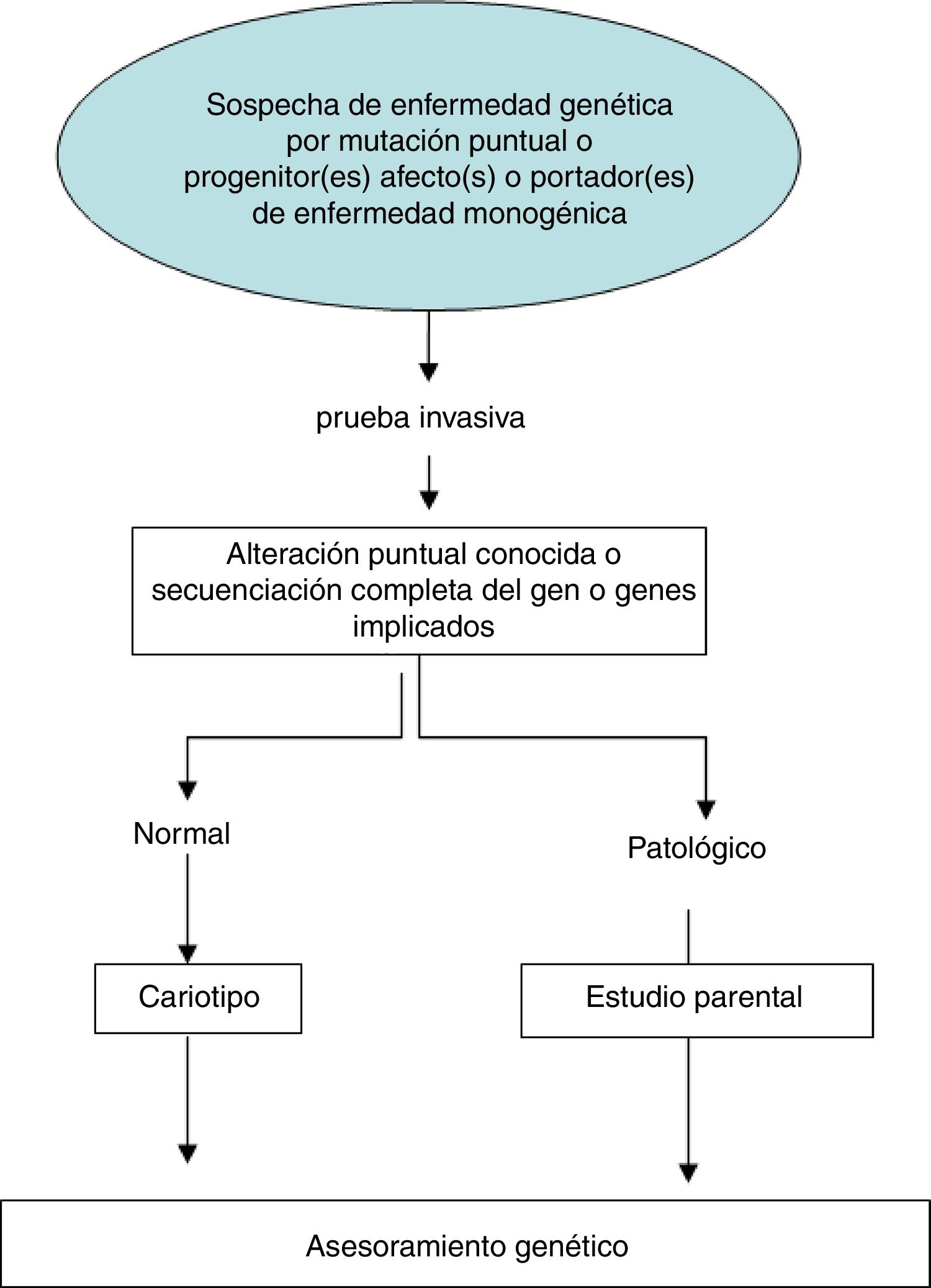
Si se sospecha alguna enfermedad genética por mutación puntual o si alguno de los progenitores está afectado o es portador de alguna enfermedad monogénica, proponemos seguir el algoritmo de la figura 5. La técnica empleada dependerá de la enfermedad que se busque, pero en la mayoría de los casos el estudio se realizará por secuenciación Sanger.
Consentimiento informadoToda actuación realizada sobre un paciente en el ámbito de la salud requiere de su consentimiento libre y voluntario una vez recibida toda la información relacionada con la misma. El consentimiento informado es crucial para preservar el principio de autonomía y los derechos de los pacientes. Normalmente, se trata de un consentimiento verbal; sin embargo, se hará por escrito en los casos en los que el paciente vaya a someterse a una intervención quirúrgica, a un procedimiento diagnóstico invasivo y en cualquier exploración que comporte riesgos o inconvenientes que puedan repercutir en la salud del paciente (Ley 41/2002 del 14 de noviembre). De acuerdo con la Orden SSI/2065/2014 (BOE del 6 de noviembre), la indicación de los análisis genéticos requiere de un consentimiento por escrito. El consentimiento informado es obligatorio y debe ser proporcionado por los profesionales de la salud, así como ser incluido en la historia clínica del paciente. En cualquier momento, la persona afectada puede revocar libremente su consentimiento.
En diagnóstico prenatal, el hecho de que las muestras generalmente utilizadas para la realización de las pruebas genéticas sean obtenidas a partir de procedimientos invasivos y que los resultados de las pruebas puedan generar importantes implicaciones sobre el futuro niño, los padres, los planes reproductivos o incluso sobre la familia, obliga a la gestante a dar su consentimiento antes de la toma de la muestra mediante un consentimiento informado por escrito41. El objetivo del consentimiento informado en el diagnóstico prenatal es proporcionar y transmitir, de forma clara, información relevante sobre la prueba diagnóstica que se ofrece a la gestante, de manera que le permita entender su contenido, la decisión que se va a tomar y sus posibles consecuencias, todo ello, en concordancia con sus necesidades individuales y sus valores. Por ello, todo consentimiento informado debe ir acompañado de un asesoramiento genético previo a la prueba, dado por un profesional cualificado, que permita a la gestante tomar decisiones informadas proporcionándole la autonomía para aceptar o no someterse a la prueba prenatal que se le ofrece. Aunque la pareja debe estar involucrada en el proceso, la última decisión recae en la madre embarazada41. Por otro lado, es responsabilidad ética de los profesionales del cuidado prenatal de preservar los principios de autonomía, beneficencia, no maleficencia y justicia del futuro niño y de la familia. Esto debe quedar reflejado en el consentimiento informado, que debe incluir los siguientes apartados41,42:
- a.
Parte informativa: informa acerca de la prueba que se ofrece.
- –
¿En qué consiste?, ¿qué estudia?, ¿cuál es su utilidad? Se debe describir cómo es la prueba, qué estudia (cromosoma, genes, etc.) y para qué enfermedades se utiliza.
- –
Propósito o indicación diagnóstica. Se debe indicar el motivo de realización de la prueba, la sospecha diagnóstica y el objetivo (identificar la alteración genética causante).
- –
Exactitud y limitaciones de la prueba. Indicar la exactitud de la prueba para la(s) enfermedad(es) a estudio, falsos positivos y falsos negativos y qué no puede detectar la prueba.
- –
Opciones alternativas a la prueba, si las hay.
- –
Beneficios: identificar la causa genética que confirma el diagnóstico de la enfermedad, además de que puede aportar información acerca del pronóstico, posibles tratamientos, etc.
- –
Posibles resultados que se pueden obtener. Cuáles se informan y cuáles no.
- 1.
Se identifica la causa de la enfermedad. A los padres se les debe explicar que esto quiere decir que el feto lleva la mutación(es) y qué efecto tendrá sobre su salud cuando nazca.
- 2.
Resultados de significado incierto. A los padres se les debe explicar que esto quiere decir que podemos encontrar resultados sobre los que no hay información previa en la literatura científica y no podemos predecir sus consecuencias.
- 3.
Hallazgos incidentales: hallazgo inesperado con potencial significado clínico, pero no relacionado con la indicación diagnóstica. A los padres se les debe explicar que esto significa que podemos encontrar mutaciones en otros genes que no estábamos buscando y que podrían tener un efecto sobre la salud del nuevo ser.
- –
Posibles implicaciones de los resultados: probabilidades de herencia, posibles implicaciones a otros miembros de la familia, penetrancia incompleta de la enfermedad causal, posibles pruebas adicionales.
- –
Tipo de muestra que se requiere para la prueba, cómo se obtiene y cuáles son sus riesgos.
- –
Tiempo dedicado a la toma de muestra y a la prueba.
- –
Planes de confidencialidad: quién tendrá acceso a la muestra y a los resultados de la prueba.
- –
Planes de conservación y custodia de la muestra.
Parte de autorización y consentimiento. En esta parte la gestante declara su conformidad, revoca o renuncia su consentimiento para la realización de la prueba. En el caso de declarar que ha entendido toda la información recibida y decide dar su consentimiento para la realización de la prueba, la gestante acepta o no acepta determinados puntos relacionados con los planes de conservación de la muestra (si desea que se destruya la muestra una vez realizada la prueba o se conserve), planes de confidencialidad (si desea o no que la muestra sea compartida de forma anónima con grupos de investigación) y los posibles resultados (si desea o no ser informado de los hallazgos incidentales o inciertos).
El consentimiento informado suele ser largo y complejo dados su carácter técnico y la cantidad de información que debe abarcar, lo que dificulta su comprensión. Además, se requiere tiempo para explicarlo adecuadamente, responder a posibles preguntas y ser entendido por la gestante. Además de la obligación legal y moral del consentimiento informado, es necesario tener en cuenta que una información insuficiente o poco clara puede dar lugar a futuras reclamaciones.
Finalmente, no solo es importante dar una información adecuada antes de la prueba, sino que además se debe ofrecer un asesoramiento genético tras la prueba que informe y explique los resultados obtenidos y proporcione apoyo a la gestante41.
La incorporación de las pruebas genéticas prenatales no invasivas ha permitido ofrecer a la gestante una prueba de cribado segura en un momento temprano del embarazo, ventajas que han supuesto un nuevo paradigma ético. Es por ello, que urge tomar parte de las cuestiones éticas que conlleva43. The National Society of Counselors (NSGC) considera las pruebas genéticas no invasivas una opción en el caso de embarazadas de alto riesgo de determinadas anomalías cromosómicas e insta a ofrecer las pruebas solo si van acompañadas de un consentimiento informado y asesoramiento por profesionales cualificados44.
Este documento tiene la conformidad de las tres Sociedades (AEBM-ML, AEFA y SEQC-ML) como Recomendación profesional en el ámbito del Laboratorio Clínico.
