





Introducción
En 1995, Meis-Kindblom y cols.5 describieron 25 casos de una variante de sarcoma cuyo particular patrón esclerosante podía simular un carcinoma. Debido a que todos los casos mostraban acúmulos de células fusiformes similares a las del fibrosarcoma convencional, los autores sugirieron que se trataba de una variante de fibrosarcoma, coincidiendo con Enzinger, que previamente la había descrito como una variante de fibrosarcoma bien diferenciado.2 Sin embargo, la extensa variedad de antígenos expresada por el tumor4,6 sugiere que la denominación de fibrosarcoma no es del todo correcta por lo que en estos momentos se ha catalogado con la denominación de fibrosarcoma epitelioide esclerosante (FEE), entendiéndose como un fibrosarcoma de bajo grado cuyo diagnostico diferencial es muy extenso.
Se presenta un caso de FEE con positividad para la vimentina y citoqueratina, con un seguimiento de 18 años que en la actualidad se encuentra clínicamente asintomático después de una recurrencia hace 9 años.
El objetivo principal de esta nota es dar a conocer la existencia de esta variante de fibrosarcoma y la importancia de reconocerlo por las implicaciones terapéuticas y pronósticas que conlleva.
Caso clínico
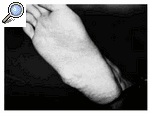
Se presenta un paciente de 40 años operado de un tumor en la planta del pie en otro hospital en 1982 a la edad de 22 años. Tenía un diámetro de 3.5 cm, un color gris-blaquecino y de consistencia dura, diagnosticado inicialmente como un fibroma juvenil aponeurótico, y descrito como una proliferación fibroblástica rodeada de un foco hialinizado. No había calcificaciones y las mitosis eran escasas. Nueve años más tarde (1991), el paciente llegó a nuestro hospital con una nueva tumoración en la misma localización (borde externo del pie derecho), de consistencia blanda y no adherida a planos profundos, con un tamaño aproximado de 2 ´3 cm. Radiológicamente no se objetivaron calcificaciones ni afectación del hueso adyacente y la ecografía describía una masa de 2.3 ´2.1 cm, sólida, heterogénea, de bordes bien definidos y lobulada, siendo ecográficamente inespecífica. Con el diagnóstico de recurrencia local (Fig. 1), se realizó extirpación en bloque del tumor, el cual tenía 4 cm de diámetro, consistencia dura y de color blanco, con áreas hemorrágicas (Fig. 2).
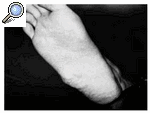
Figura 1. Tumoración en la planta del pie.

Figura 2. Imagen macroscópica del tumor, con la superficie lobulada y color blanco.
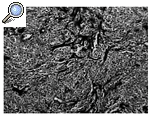
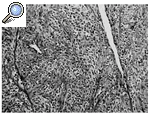
Histológicamente el tumor, que tenía un margen lobular bien definido, mostró una celularidad variable, alternando zonas con una gran celularidad con otras muy hialinizadas. Las células se organizaban en cordones, entramados y racimos o aisladas rodeadas por colágeno (Figs. 3 y 4); tenían una forma ovoidea o fusiforme y una moderada cantidad de citoplasma claro y ligeramente eosinofílico. El núcleo era pequeño, pálido o vesicular con nucleolos poco llamativos y escaso pleomorfismo. Las mitosis aparecían en un número de 4/10 campos de gran aumento (Fig. 5). No se apreciaban áreas de necrosis y existía invasión del espacio vascular. El estudio inmunohistoquímico mostraba expresión por las células neoplásicas de vimentina de modo masivo y en un elevado número de células de citoqueratina de bajo peso molecular (CAM 5,2). Eran negativas para otras queratinas (AE1, AE3, CK8), antígeno de membrana epitelial (EMA), desmina, actina y proteína S-100.
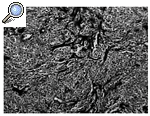
Figura 3. FEE caracterizado por abundante tejido colágeno hialinizado entre pequeñas células redondas con citoplasma claro (HE, 100x).
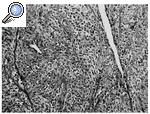
Figura 4. Otra zona del tumor. Nidos y cordones de células con esclerosis (HE, 100x).

Figura 5. Nidos de células pequeñas y ovoides con citoplasma claro. Se aprecia una imagen de mitosis (HE, 400x).
El diagnostico final fue de un sarcoma sinovial fibroso monofásico grado I. El paciente rechazó la amputación y la quimioterapia coadyuvante. Nueve años más tarde se encuentra clínicamente asintomático y debido a la revisión del caso de acuerdo con las últimas publicaciones se ha clasificado actualmente como FEE.
Discusión
El diagnóstico de los tumores de partes blandas es todavía difícil y supone un desafío. Dentro de ellos, el fibrosarcoma supone del 5 al 12% de los tumores malignos de partes blandas, afectando a personas de cualquier edad aunque más frecuentemente entre los 30 y los 70 años.1 Se pueden desarrollar de novo sobre tejido sano y también en áreas de cicatrización o traumatismos previos. El 30-60% de los fibrosarcomas se encuentran en la extremidad inferior, pero sólo el 2.5% se localizan en la zona del pie y tobillo.1 La recurrencia local varía del 29 al 60% después de una cirugía radical, pero este índice aumenta hasta el 90% si sólo se realiza una resección marginal.1
En los primeros estudios de neoplasias de tejidos blandos el diagnóstico de fibrosarcoma fue realizado en exceso. Actualmente, con el reconocimiento de la proliferación fibrosa benigna y de otros tipos de sarcoma como los tumores malignos de la vaina de nervios periféricos, los sarcomas sinoviales o los mesoteliomas fibrosos, el diagnóstico de fibrosarcoma es de «exclusión» y probablemente se produce con menor frecuencia de la que realmente le corresponde.3 También contribuye a ello el abandono de las técnicas ultraestructurales tras el uso de la inmuno-histoquímica y de hecho, éstas no siempre ofrecen un diagnóstico correcto e incluso pueden inducir a error.
El caso que se presenta tiene una descripción histológica muy parecida a la de la serie publicada por Meis-Kindblom y cols. en 19955 y que denominaron fibrosarcoma epitelioide esclerosante. Fue diagnosticado en un primer momento como una lesión benigna, un fibroma juvenil aponeurótico, debido al escaso número de mitosis y atipias celulares siendo imposible asegurar con los datos que disponíamos, si se trataba de un fibrosarcoma de bajo grado inicialmente o representaba una transformación maligna de un fibroma, aunque se sabe que dicha transformación es muy infrecuente; posteriormente, la recidiva 9 años después fue interpretada como sarcoma sinovial monofásico fusocelular grado I y por último, la buena evolución clínica del paciente y la descripción de nuevas variantes anatomopatológicas hizo que se reclasificase como FEE.
El diagnóstico diferencial es muy amplio y debe de tenerse en cuenta tanto lesiones benignas del tipo de fibromatosis, histiocitoma fibroso, fascitis nodular, miositis osificante, leiomioma hialinizante o tumor desmoide, como lesiones malignas incluyendo en estas últimas los carcinomas metastásicos, el linfoma y el sarcoma sinovial con todas sus variantes.2,7,8,9
El FEE es un sarcoma de crecimiento relativamente lento y de bajo grado de malignidad, que puede ser tratado con una extirpación quirúrgica adecuada, pero que es capaz de producir metástasis a distancia, a veces muchos años después del tratamiento de la lesión primaria,6 de ahí la importancia de su diagnóstico correcto y su diferenciación de otras lesiones benignas y, sobre todo, de aquellas malignas más agresivas. El tiempo de recurrencia referido en la serie de Meis y Kindblom se establece en 4,8 años,5 sin embargo en nuestro caso ha sido de 9 años. Ello nos obliga a realizar un seguimiento prolongado de todos los pacientes con este tipo de tumor.
Hasta que se puedan hacer más estudios con microscopía electrónica, los autores coinciden con Meis-Kindblom5 en que el diagnóstico de FEE se puede hacer por su patrón histológico, a pesar de su anómalo inmunofenotipo para ser un fibrosarcoma, siendo extremadamente cautos en la interpretación de los hallazgos inmuno-histoquímicos.
