



La célula perivascular epiteloide (PEC) es un tipo celular constante presente en un grupo de tumores que incluyen el angiomiolipoma, tumores «de azúcar» de células claras pulmonares y de sitios extrapulmonares, linfangioleiomiomatosis, entre otros. Las características de la PEC incluyen: apariencia epiteloide con citoplasma claro agranular, un núcleo central redondo a oval y un nucléolo discreto además de expresar marcadores inmunohistoquímicos únicos. Únicamente han sido descritos 11 casos de presentación ósea primaria desde su primer reporte en 2002.
ObjetivoPresentar el caso de un tumor de células perivasculares epiteloides óseo primario.
Reporte de casoVarón de 24 años de edad con dolor de un año de evolución y lesión lítica de tibia proximal derecha y extensión a partes blandas. Diagnóstico histológico de tumor de células perivasculares epiteloides óseo e inmunohistoquímica negativa.
ResultadosSeguimiento de 2 años después del tratamiento con quimioterapia adyuvante (epirrubicina/cisplatino) y de la resección en bloque; el paciente se encuentra libre de enfermedad.
ConclusionesEste es el primer caso de tumor de células perivasculares epiteloides óseo primario reportado en Latinoamérica. No encontramos los marcadores inmunohistoquímicos y creemos que esto puede deberse a variaciones étnicas no descritas.
Perivascular epithelioid cell (PEC) is a cell type constantly present in a group of tumours including angiomyolipoma (AML), clear-cell «sugar» tumour (CCST) of the lung and extrapulmonary sites, lymphangioleiomyomatosis (LAM), and clear-cell tumours of other anatomical sites. It has morphologic distinctive features: epithelioid appearance with a clear to granular cytoplasm, a round to oval, centrally located nucleus and an inconspicuous nucleolus. Immunohistochemically, PEC expresses myogenic and melanocytic markers. Eleven cases of primary bone PEComa presentation have been described since 2002.
ObjectiveTo report a case of primary bone perivascular epithelioid cell tumour.
Case report24 year-old male presented with pain. X-ray revealed an osteolytic lesion at right proximal tibia with soft tissue extension. Evaluation of slides identified a bony perivascular epithelioid cell tumour without immunohistochemical study confirmation.
ResultsPatient was treated by surgical excision and adjuvant chemotherapy (epirubicin/cysplatin). After two years of follow-up the patient remains disease free.
ConclusionsThis is the first-case report in Latin America. Immunohistochemical stains were negative and we believe it may be due to non-described ethnic variations.
La célula perivascular epiteloide (PEC) es un tipo celular constante presente en un grupo de tumores que incluyen el angiomiolipoma (AML), tumores «de azúcar» de células claras pulmonares y de sitios extrapulmonares, linfangioleiomiomatosis, tumor melanocítico de células claras del ligamento falciforme/ligamento redondo y tumores raros de células claras de otros sitios anatómicos1.
Las PEC fueron descritas por primera vez por Apitz en 19432; y Masson las reportó como un «mioblasto anormal» en AML renal en su libro3. Sin embargo, el término «célula perivascular epiteloide» fue acuñado por Bonetti et al. en referencia a lesiones epiteloides con citoplasma claro/acidófilo y distribución perivascular4.
Las características distintivas de la PEC incluyen la apariencia epiteloide con citoplasma claro agranular, un núcleo central redondo a oval y un nucléolo discreto. Esta exhibe marcadores inmunohistoquímicos miogénicos y melanocíticos tales como HMB45, HMSA-1, MelanA/Mart 1, Mitf, actina y raramente desmina y se relacionan con esclerosis tuberosa. Existen dudas acerca de la histogénesis de los tumores de células perivasculares epiteloides de la definición de AML epiteloide y de la identificación de criterios histológicos de malignidad1,5.
La Organización Mundial de la Salud ha definido el tumor de células perivasculares epiteloides (PEComa) como un «tumor mesenquimatoso compuesto de células perivasculares epiteloides con características histológicas e inmunohistoquímicas distintivas». Actualmente el PEComa es una entidad ampliamente aceptada. Sin embargo, algunos autores dudan de la existencia del PEComa como un tumor distintivo1.
Los PEComas son considerados tumores ubicuos y han sido descritos en diferentes órganos, tales como: riñón, vejiga, próstata, útero, ovario, vulva, vagina, pulmón, páncreas e hígado mayoritariamente. Raramente se han descrito en el hueso, entre otros sitios anatómicos menos frecuentes1.
Caso clínicoSe obtuvo el consentimiento informado del paciente y no se cuenta con datos que hagan capaz la identificación del sujeto.
Varón de 24 años de edad con diagnóstico histológico de PEComa, manifestado por dolor y aumento de volumen en la tibia proximal derecha de crecimiento progresivo, de un año de evolución. El paciente no tuvo historia médica familiar o personal con antecedentes de importancia, y los exámenes de laboratorio se encontraron en los límites normales. No se identificaron alteraciones de metabolismo del calcio.
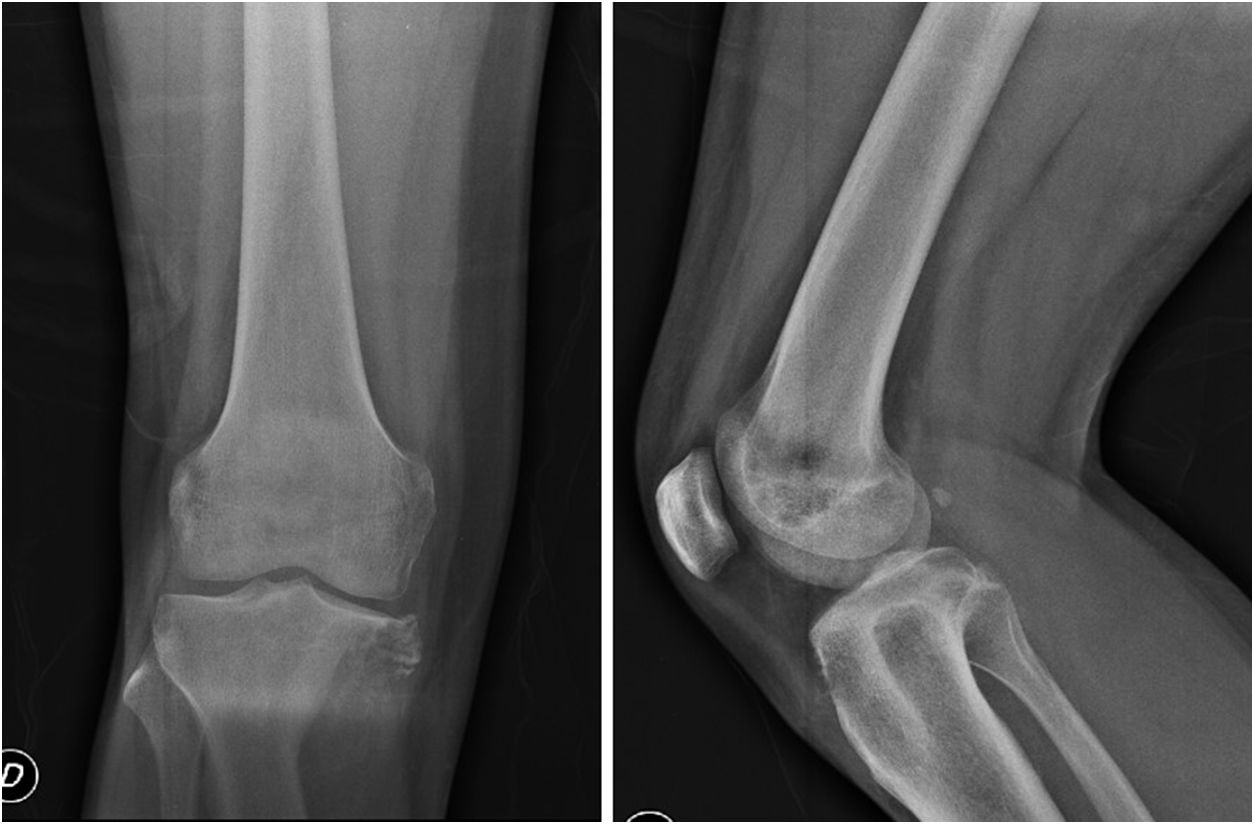
En las radiografías convencionales (fig. 1) se encuentra una lesión lítica epifisiometafisaria de la tibia proximal derecha, sin reacción perióstica y con extensión a partes blandas en la región anteromedial y posterior de la pierna.

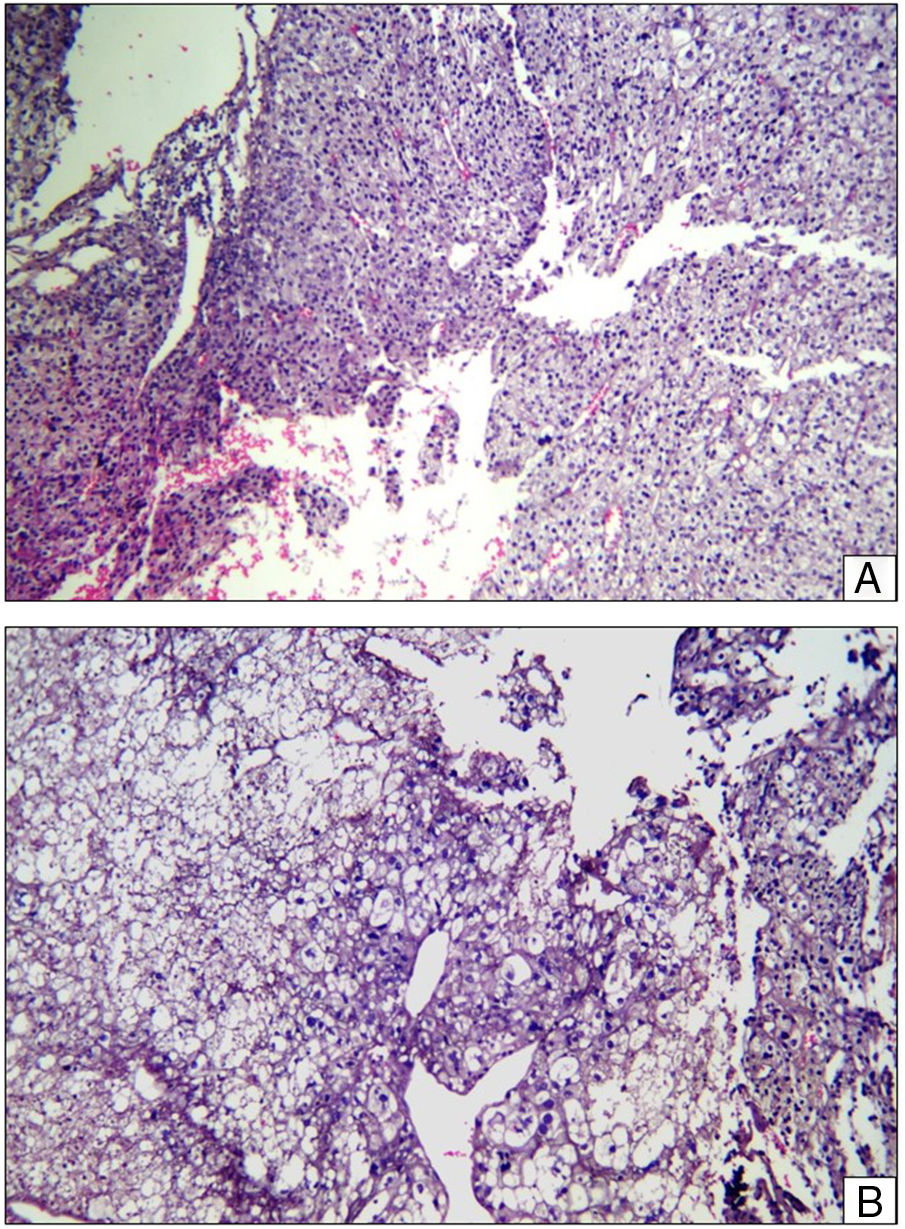
Como parte del protocolo de estudio en el paciente con sospecha de tumor óseo se realizó biopsia incisional, donde se reportó la presencia de neoplasia maligna de células claras de aspecto epiteloide cohesivas dispuestas alrededor de los vasos, con citoplasma claro granular y nucléolo prominente (morfología de PEComa) (fig. 2). El estudio de inmunohistoquímica reportó: citoqueratina 8/18 (–), antígeno de membrana epitelial (–), HMB 45 (–), PAX-2 (–), antígeno de carcinoma renal (–), actina de músculo liso (–), factor de transcripción de microftalmia (–), S-100 (–), CD 99 (–), Melan A (–), PAX-8 (–), CD 117 (–).

Corte histopatológico. A. Se observan mantos de células epitelioides con citoplasma de predominio granular, núcleo central hipercromático y nucléolo prominente alrededor de espacios vasculares. Áreas de hemorragia. Hematoxilina-eosina×20. B. Se observan mantos de células epitelioides de citoplasma claro y en algunas vacuolado, con nucleo hipercromático central, alrededor de los espacios vasculares. Hematoxilina-eosina×40.
Fuente: archivo clínico.
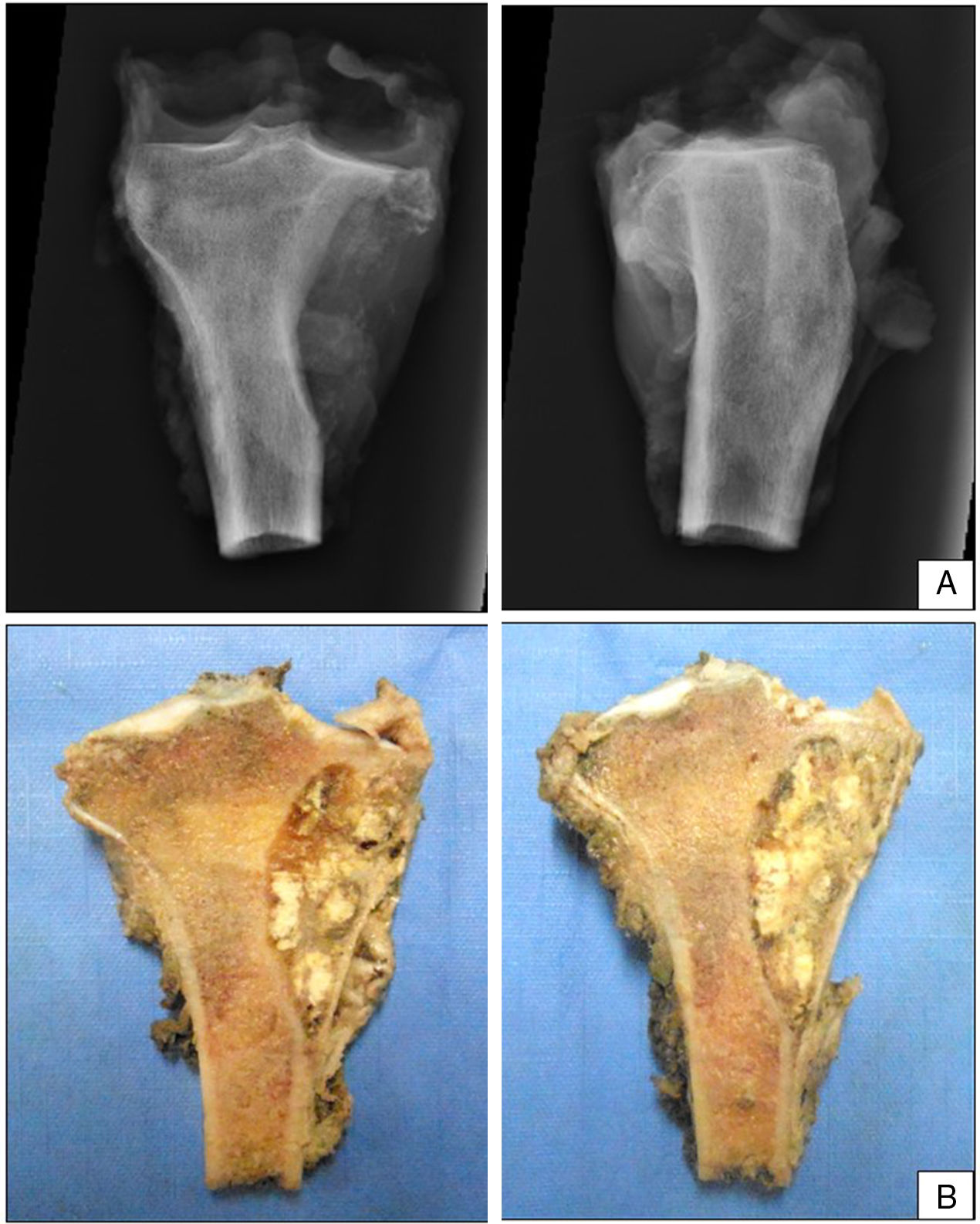
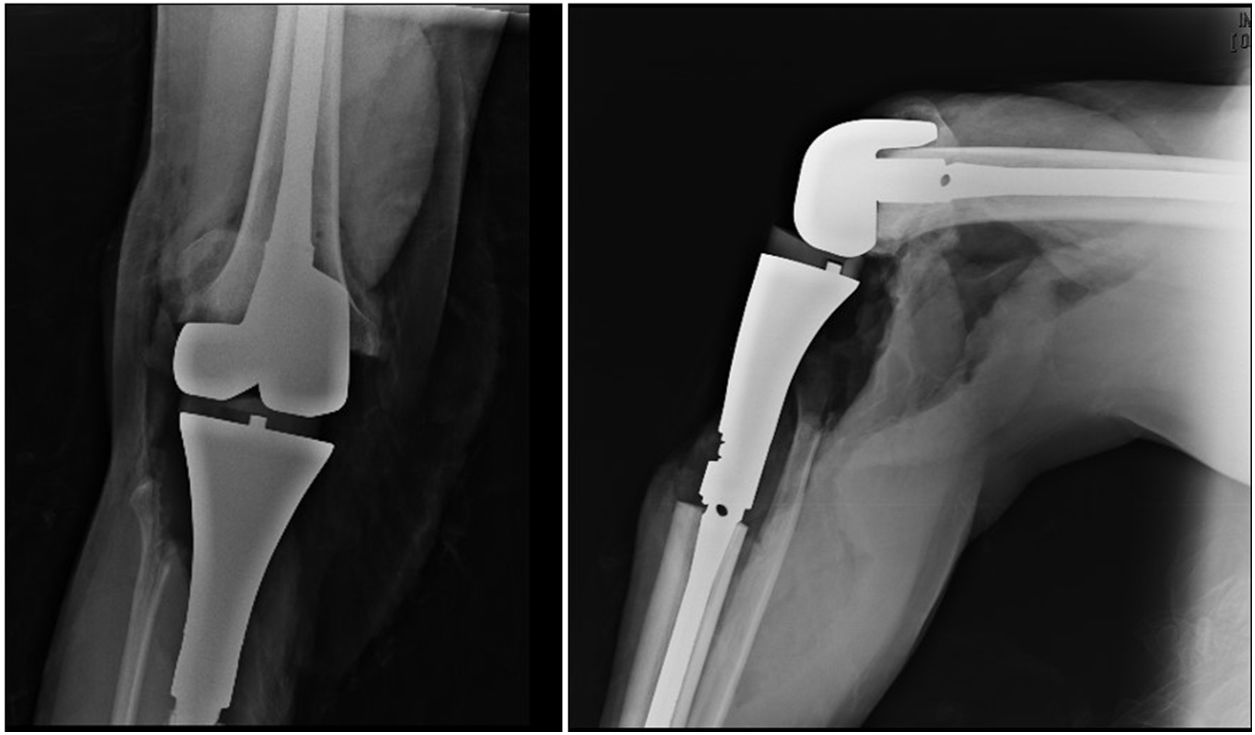
El paciente siguió con tratamiento de quimioterapia neoadyuvante con epirrubicina/cisplatino por 3 ciclos con adecuada respuesta (necrosis tumoral>90%), por lo que se realizó resección en bloque de la tibia proximal con márgenes amplios (fig. 3) y reconstrucción esquelética con prótesis tumoral (GMRS Stryker proximal tibia, cementada, Mahwah, NJ) (fig. 4). El estudio patológico de la pieza quirúrgica reportó necrosis tumoral del 98% y cambios posquimioterapia.


A los 24 meses de seguimiento el paciente continúa vivo, sin datos de enfermedad tumoral y sin complicaciones agregadas.
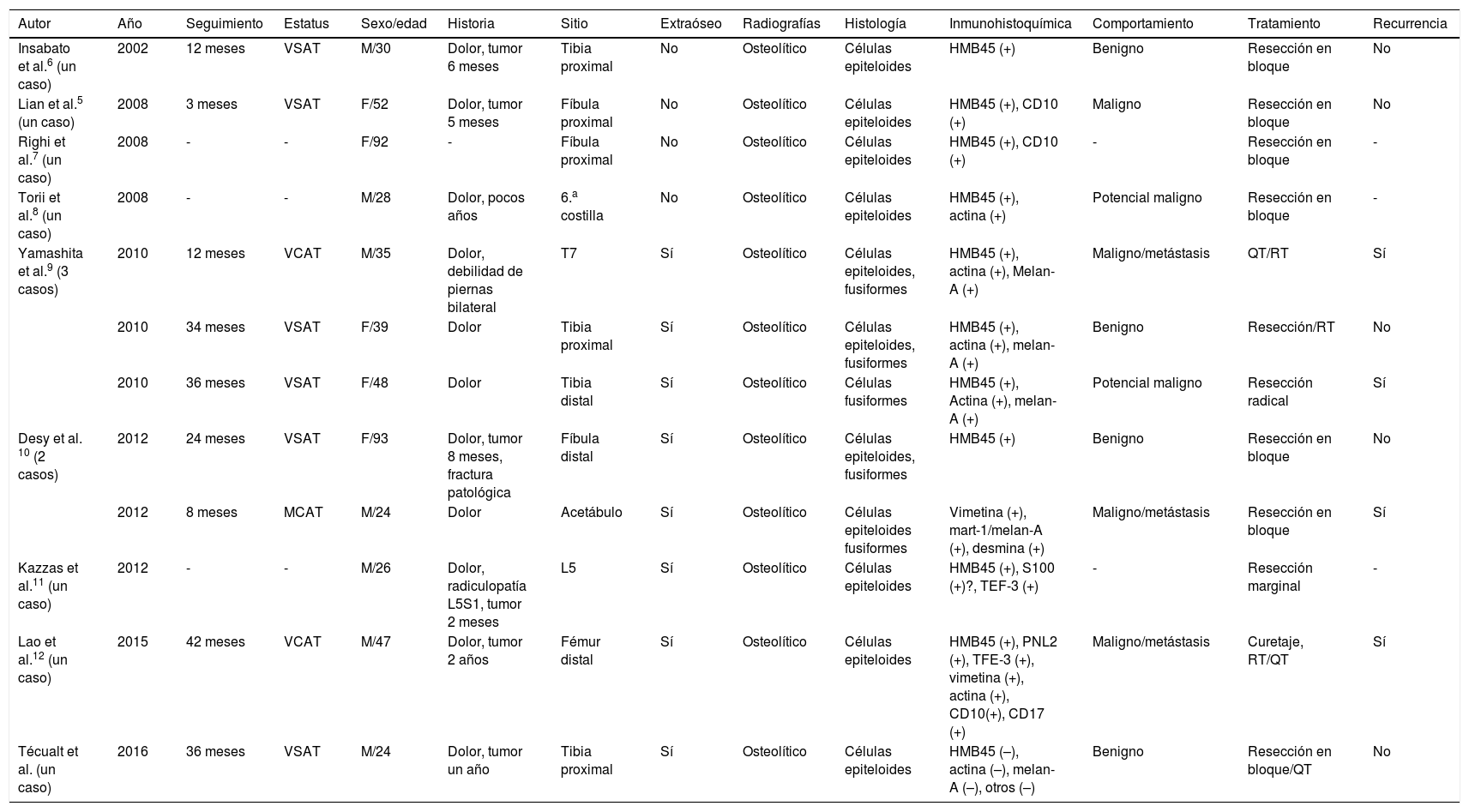
DiscusiónEl PEComa primario óseo no es común, y se han reportado pocos en la literatura5. En la tabla 1 se enumeran los casos que han sido reportados hasta el momento5–12.
Características clínicas de 12 PEComa óseos primarios
| Autor | Año | Seguimiento | Estatus | Sexo/edad | Historia | Sitio | Extraóseo | Radiografías | Histología | Inmunohistoquímica | Comportamiento | Tratamiento | Recurrencia |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Insabato et al.6 (un caso) | 2002 | 12 meses | VSAT | M/30 | Dolor, tumor 6 meses | Tibia proximal | No | Osteolítico | Células epiteloides | HMB45 (+) | Benigno | Resección en bloque | No |
| Lian et al.5 (un caso) | 2008 | 3 meses | VSAT | F/52 | Dolor, tumor 5 meses | Fíbula proximal | No | Osteolítico | Células epiteloides | HMB45 (+), CD10 (+) | Maligno | Resección en bloque | No |
| Righi et al.7 (un caso) | 2008 | - | - | F/92 | - | Fíbula proximal | No | Osteolítico | Células epiteloides | HMB45 (+), CD10 (+) | - | Resección en bloque | - |
| Torii et al.8 (un caso) | 2008 | - | - | M/28 | Dolor, pocos años | 6.a costilla | No | Osteolítico | Células epiteloides | HMB45 (+), actina (+) | Potencial maligno | Resección en bloque | - |
| Yamashita et al.9 (3 casos) | 2010 | 12 meses | VCAT | M/35 | Dolor, debilidad de piernas bilateral | T7 | Sí | Osteolítico | Células epiteloides, fusiformes | HMB45 (+), actina (+), Melan-A (+) | Maligno/metástasis | QT/RT | Sí |
| 2010 | 34 meses | VSAT | F/39 | Dolor | Tibia proximal | Sí | Osteolítico | Células epiteloides, fusiformes | HMB45 (+), actina (+), melan-A (+) | Benigno | Resección/RT | No | |
| 2010 | 36 meses | VSAT | F/48 | Dolor | Tibia distal | Sí | Osteolítico | Células fusiformes | HMB45 (+), Actina (+), melan-A (+) | Potencial maligno | Resección radical | Sí | |
| Desy et al. 10 (2 casos) | 2012 | 24 meses | VSAT | F/93 | Dolor, tumor 8 meses, fractura patológica | Fíbula distal | Sí | Osteolítico | Células epiteloides, fusiformes | HMB45 (+) | Benigno | Resección en bloque | No |
| 2012 | 8 meses | MCAT | M/24 | Dolor | Acetábulo | Sí | Osteolítico | Células epiteloides fusiformes | Vimetina (+), mart-1/melan-A (+), desmina (+) | Maligno/metástasis | Resección en bloque | Sí | |
| Kazzas et al.11 (un caso) | 2012 | - | - | M/26 | Dolor, radiculopatía L5S1, tumor 2 meses | L5 | Sí | Osteolítico | Células epiteloides | HMB45 (+), S100 (+)?, TEF-3 (+) | - | Resección marginal | - |
| Lao et al.12 (un caso) | 2015 | 42 meses | VCAT | M/47 | Dolor, tumor 2 años | Fémur distal | Sí | Osteolítico | Células epiteloides | HMB45 (+), PNL2 (+), TFE-3 (+), vimetina (+), actina (+), CD10(+), CD17 (+) | Maligno/metástasis | Curetaje, RT/QT | Sí |
| Técualt et al. (un caso) | 2016 | 36 meses | VSAT | M/24 | Dolor, tumor un año | Tibia proximal | Sí | Osteolítico | Células epiteloides | HMB45 (–), actina (–), melan-A (–), otros (–) | Benigno | Resección en bloque/QT | No |
MCAT: muerto con actividad tumoral; QT: quimioterapia; RT: radioterapia; VCAT: vivo con actividad tumoral; VSAT: vivo sin actividad tumoral.
Nosotros reportamos el caso número 12 de PEComa primario óseo desde la primera descripción hecha por Insabato et al. en 20026, y el tercero en la tibia proximal. Este tiende a involucrar los huesos largos de las extremidades en esta presentación, y el dolor es la característica de presentación más común, seguido por fractura patológica y tumor9, los cuales han sido encontrados igualmente en nuestro paciente.
El caso que nosotros reportamos es consistente con el sitio de afección, las características histopatológicas clásicas descritas para el PEComa: células claras epiteloides con citoplasma claro granular, nucléolo prominente y disposición de estas células alrededor de los vasos. Sin embargo, el reporte de inmunohistoquímica no arrojó resultados positivos para los marcadores habitualmente encontrados en esta estirpe tumoral: MelanA, actina de músculo liso y HMB-45.
La histogénesis y la contraparte normal/fisiológica de las PEC son desconocidas, pero se han propuesto algunas hipótesis. Una de estas hipótesis es que las PEC derivan de células no diferenciadas de la cresta neural que expresan el fenotipo dual de músculo liso y melanocítico; una segunda hipótesis es que las PEC tienen un origen mioblástico, muscular liso con una alteración molecular que provoca la expresión de melanogénesis y marcadores melanocíticos; una tercera hipótesis es que las PEC tienen un origen pericítico1. Por otro lado, al menos focalmente, las células tumorales de PEComa se ordenan estrechamente alrededor de los vasos, y en algunas ocasiones parecen comprometer la pared muscular de los vasos sanguíneos9.
Folpe et al. han propuesto la clasificación provisional del PEComa en «benigno», «potencial maligno incierto» y «maligno». Una asociación significativa fue encontrada entre el tamaño del tumor mayor a 5cm, patrón de crecimiento infiltrante, alto grado nuclear, alta celularidad, necrosis y actividad mitótica mayor de 1/50 por campo de alto poder. Se ha sugerido que los PEComas, teniendo 2 o más de estas características preocupantes, pueden ser clasificados como «malignos», reconociendo que la enfermedad clínicamente agresiva puede no ser vista en todas las neoplasias malignas histológicamente. En nuestro caso se han cumplido los criterios descritos por Folpe et al. También ha sido sugerido que los tumores que muestran únicamente una de estas características pueden ser clasificados como teniendo un «potencial maligno incierto»13.
El PEComa maligno puede ser una enfermedad muy agresiva, llevando a múltiples metástasis y muerte, tal como se espera en los sarcomas de alto grado, y existen reportes de que los PEComas malignos han metastatizado después de 7 a 9 años en sitios de afección primaria distintos al hueso1.
Actualmente parece que el único abordaje en el tratamiento de los PEComas en los casos agresivos es la cirugía, ya que la radio y quimioterapia no han mostrado resultados significativos. Sin embargo, esta información se deriva de casos anecdóticos y no se han implementado ensayos terapéuticos. Una de las dificultades obvias para llevar al cabo este tipo de estudios es la rareza de la enfermedad1.
En nuestro caso se ha utilizado quimioterapia neoadyuvante debido a la poca experiencia local que se tiene en el tratamiento de este tipo de lesiones, puesto que es el primero que se ha encontrado en nuestro país. Sin embargo, puesto que se reportó por parte de patología cambios sugestivos de un probable sarcoma y atipia, se decidió iniciar el tratamiento con esta modalidad de terapia. Una vez revisado el estudio histopatólogico por el patólogo experto en tejido óseo no se encontró similitud alguna con otro tipo de sarcomas, y fue hasta el reporte posterior a la resección de la pieza quirúrgica cuando se ha dado el diagnóstico definitivo de PEComa una vez que se ha complementado su estudio con inmunohistoquímica.
El pronóstico del PEComa es variable y depende primariamente de las características histopatológicas10.
ConclusionesEste es el primer caso de PEComa óseo primario reportado en Latinoamérica. Se demostraron sus características clinicopatológicas, sin embargo no se encontraron los marcadores de inmunohistoquímica, y este último punto no concuerda con lo clásicamente descrito. Creemos que esto puede deberse a variaciones étnicas hasta este momento no descritas.
Nivel de evidenciaNivel de evidencia V.
