




La enfermedad de Creutzfeldt-Jakob esporádica (ECJe) forma parte de las enfermedades priónicas, caracterizadas por largos periodos de incubación y progresión inexorable una vez aparecidos los síntomas1, con una incidencia anual de un caso por millón de habitantes2.
Presentamos el caso de una mujer de 77 años, con evolución atípica de ECJe y supervivencia mucho mayor a la habitual. Fue remitida por su médico de Atención Primaria al equipo de Atención Geriátrica Domiciliaria del Hospital Cruz Roja por deterioro funcional severo tras ingreso hospitalario.
Sin antecedentes relevantes, en marzo de 2014 había sido diagnosticada de probable ECJe. Comenzó con cefalea y mínima claudicación en hemicuerpo izquierdo, estudiándose mediante tomografía computerizada cerebral, eco-Doppler de troncos supraaórticos, electroencefalograma y líquido cefalorraquídeo con proteína 14-3-3, todo ello sin alteraciones, y resonancia magnética (RM) cerebral con lesiones sugestivas de encefalopatía espongiforme. Al año fue ingresada por deterioro neurológico progresivo: disgrafía, dislexia, bradipsiquia, síntomas depresivos, conservando comprensión, repetición y nominación, sin mioclonías. Se repitieron las mismas pruebas complementarias, estudio genético del gen PRPN, con similares resultados (proteína 14-3-3 negativa en líquido cefalorraquídeo), salvo electroencefalograma donde aparecieron lesiones típicas, confirmando diagnóstico de probable ECJe.
La paciente mantenía situación funcional de independencia en actividades básicas de la vida diaria salvo baño y vestido, deambulación autónoma en domicilio, ocasionalmente bastón, e incontinencia urinaria ocasional (Escala de Cruz Roja Funcional 1-2/5, índice de Barthel modificado 83/100). Mentalmente presentaba desorientación temporal, mantenía conversaciones sencillas (Escala de Cruz Roja Mental 2/5). Buen apoyo familiar, con esposo y cuidadora 24 h.
Hasta julio de 2016, que sufrió ingreso hospitalario por infección urinaria. Fue remitida tras el alta al equipo de Atención Geriátrica Domiciliaria, apreciándose deterioro funcional severo, dependencia para todas las actividades básicas de la vida diaria (Cruz Roja Funcional 5/5, índice de Barthel modificado 0/100), labilidad emocional, quejidos constantes, inquietud psicomotriz nocturna e insomnio con somnolencia diurna. Había acudido al Servicio de Urgencias, pautándose haloperidol y bromazepam, sin mejoría. Referían anorexia, dificultad para tragar y dolor abdominal. En la inspección de cavidad oral se apreciaba gran sequedad de mucosas, secreciones adheridas y placas blanquecinas en paladar compatibles con candidiasis oral.
Se trató con nistatina oral y recomendaciones de higiene bucal. Para control del sueño-vigilia y alteraciones psicoconductuales se iniciaron mirtazapina15mg y lorazepam 1mg nocturnos, suspendiendo haloperidol y bromazepam. Se detectó por urocultivo una infección de tracto urinario, tratándose según antibiograma. Presentó mejoría clínica franca del estado general, recuperó apetito e inició una ingesta alimentaria adecuada. Se controló el sueño, desapareció la inquietud psicomotriz, mejoró anímicamente y mantenía conversación sencilla. Se recomendaron ejercicios de activación muscular consiguiendo deambulación con ayuda de una persona en domicilio, colaboración en actividades básicas de la vida diaria y continencia diurna.
Inevitablemente, durante el seguimiento por Atención Geriátrica Domiciliaria progresaron lentamente los síntomas neurológicos: al año, empeoraron los trastornos del lenguaje (nominación y repetición) y aparecieron mioclonías. En febrero de 2018, cuando falleció, casi 4 años después del inicio de la sintomatología, presentaba deterioro funcional severo, mutismo, mirada perdida, y frecuentes mioclonías en cabeza y extremidades.
DiscusiónLas enfermedades priónicas son procesos neurodegenerativos caracterizados por pérdida neural y presencia de vacuolas que dan aspecto espongiforme3, por eso también se conocen como encefalopatías espongiformes transmisibles (EET), y acumulación neuronal de isoformas patológicas de la proteína priónica (PrPSc). La tríada característica es la combinación de deterioro cognitivo, síntomas psiquiátricos y alteraciones motoras.
Se pueden clasificar etiológicamente como: EET adquirida (< 5% de los casos), EET familiar (10-15%) y EET idiopáticas o esporádicas (85%). Dentro de estas últimas, la más frecuente es la ECJe.
En la ECJe pueden diferenciarse subtipos basándose en el polimorfismo del codón 129 del gen PRPN (para el aminoácido valina V o metionina M4), que codifica la PrPSc, y el peso molecular de la PRPSc (tipo 1 o 2)2,3. Según esto existen 5 subtipos (MM1/MV1, MM2, VV1, VV2, MV2), con clínica y supervivencias diferentes, que varía entre 4 y 17 meses, falleciendo el 85-90% de los pacientes antes del año3.
La edad media de inicio son 67 años3. Su presentación inicial es muy variable, dificultando y retrasando el diagnóstico5. Los síntomas prodrómicos son inespecíficos: pérdida de peso, alteraciones del sueño y de conducta, y síntomas visuales4, afectándose la marcha y el lenguaje en fases tempranas. Posteriormente aparece una demencia rápidamente progresiva acompañada habitualmente de ataxia cerebelosa y mioclonías, progresando a mutismo acinético4.
Los criterios diagnósticos más aceptados son los propuestos por Zerr et al.6 (sensibilidad 98%) que incluyen: 1) demencia progresiva; 2) al menos uno de estos síntomas: mioclonías, piramidalismo o extrapiramidalismo, disfunción cerebelar/visual, mutismo acinético; 3) electroencefalograma típico (complejos periódicos sobre actividad de fondo enlentecida con ondas agudas bifásicas/trifásicas con frecuencia de 0,5-2Hz) con elevación de proteína 14-3-3 en líquido cefalorraquídeo; o bien, imagen típica de RM cerebral (hiperintensidad de señal en frecuencias FLAIR y difusión en estriado y/o corteza cerebral) y 4) descartar otras posibles causas.
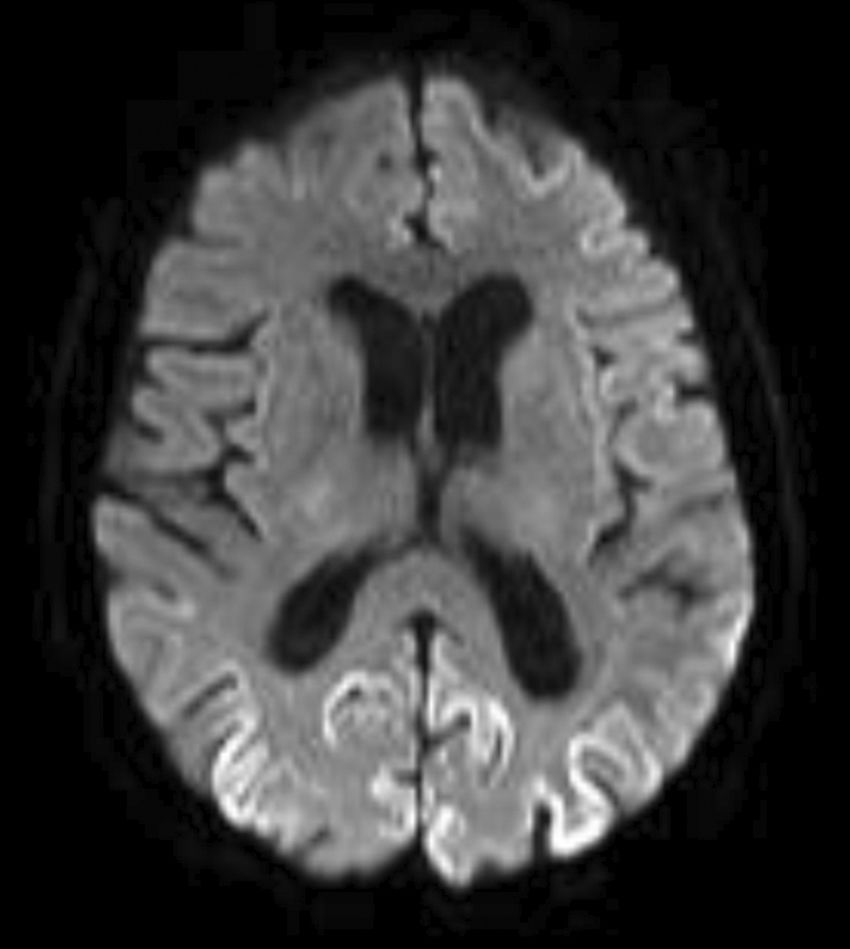
El diagnóstico definitivo es con confirmación patológica tras biopsia cerebral. La prueba complementaria con mayor superioridad diagnóstica es la RM cerebral, principalmente las secuencias FLAIR y DWI, que usadas conjuntamente elevan la sensibilidad y especificidad (91 y 95% respectivamente)3. Como en nuestra paciente, que presentaba hiperintensidad en circunvoluciones parietales posteriores y occipitales (fig. 1). Tras el estudio genético del gen PRPN, con heterocigosis en codón 129 metionina/valina (129M/V), sin mutación, el diagnóstico fue de ECJeMV2, con una supervivencia media de 17 meses según la literatura5.
 que muestra la restricción de la difusión con hiperintensidad de la señal giriforme de distribución parcheada en circunvoluciones parietales posteriores y occipitales, compatible con encefalopatía espongiforme.")
Corte axial de la resonancia magnética cerebral en secuencia de difusión (DWI) que muestra la restricción de la difusión con hiperintensidad de la señal giriforme de distribución parcheada en circunvoluciones parietales posteriores y occipitales, compatible con encefalopatía espongiforme.
Existen múltiples presentaciones de las enfermedades priónicas, que a pesar de los avances diagnósticos, continúan siendo relativamente desconocidas, sobre todo en relación con sintomatología, supervivencia y evolución atípicas. Esto supone un reto para su manejo y necesidad de un pronóstico que permita establecer un plan de cuidados adecuado. La paciente descrita presentó un inicio tardío de la sintomatología y una supervivencia muy superior a la referida en la bibliografía. La valoración geriátrica permitió el manejo domiciliario de algunos de sus síntomas, no atribuibles a su ECJe, contribuyendo a prolongar su supervivencia y aportando una mayor calidad de vida a la paciente y sus cuidadores.
Al doctor Juan Ignacio González Montalvo por su revisión crítica del manuscrito.
