




La micosis fungoide (MF) es un linfoma primitivamente cutáneo derivado de la proliferación neoplásica de linfocitos T cooperadores. Aunque el proceso se origina en la piel y permanece como un proceso, solo cutáneo, durante periodos prolongados de tiempo, en fases avanzadas de su evolución puede extenderse a ganglios linfáticos y vísceras internas1. Presentamos un singular caso de MF en fase tumoral que evoluciona de forma excepcional a un linfoma no Hodgkin.
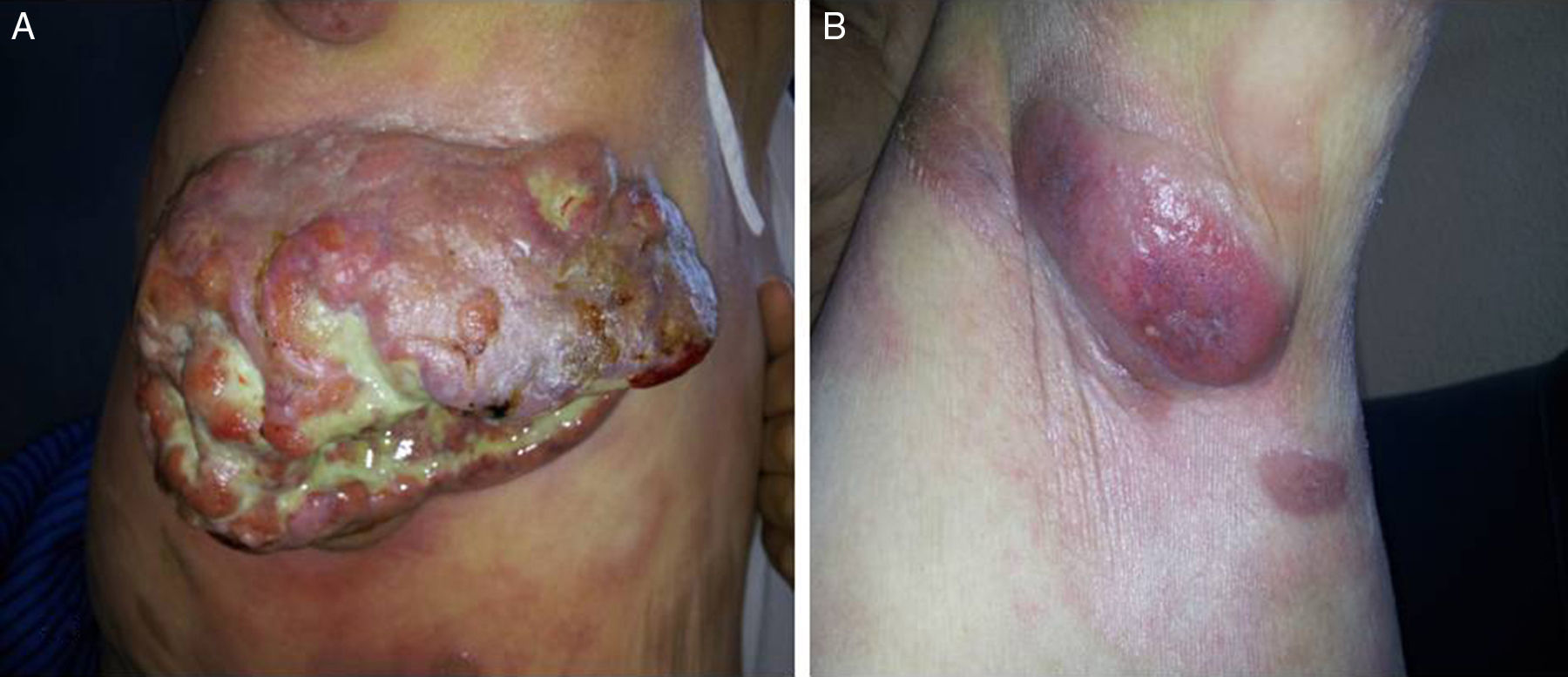
Mujer de 76 años, sin antecedentes médicos de interés. Valoración geriátrica integral: a) escala de Lawton-Brody para actividades instrumentales: 8 (rango de puntuación: 0-8); b) test de Pfeiffer: 2 errores (rango de puntuación: 0-10); c) escala de Yesavage: 5 (rango de puntuación: 0-15), y d) escala de valoración socio-familiar de Gijón: 7 (rango de puntuación: 5-25). Consultó por gran tumoración indolora en región axilar derecha de 6 meses de evolución, así como lesiones cutáneas eritematosas asintomáticas de reciente aparición de distribución universal, sin asociar sintomatología sistémica. En la exploración destacaba una tumoración axilar derecha ulcerada de 15×10cm con signos de necrosis y sobreinfección. En axila izquierda se apreciaba inicio de tumoración inflamatoria de menor tamaño (fig. 1). Del mismo modo presentaba placas eritemato-descamativas en zonas no fotoexpuestas: espalda, axilas, abdomen y proximal de extremidades. El resto de la exploración era rigurosamente normal. En el estudio analítico destacaba hemograma, hemostasia, perfil renal, lipídico, tiroideo, ferrocinético y hepático, vitamina B12, folato y sedimento urinario dentro de la normalidad. Las serologías a CMV, VHB, VHC y a VIH eran negativas a excepción de VEB IgG positivo. ß-2 microglobulina y PCR presentaban también valores normales. El frotis de sangre periférica reflejaba un 4-5% de células linfoides de pequeño tamaño con núcleos irregulares o de aspecto cerebroide. La TAC de tórax-abdomen describía la lesión cutánea axilar derecha con adenopatías locales menor de 1cm sin otra afectación orgánica. El cultivo del exudado axilar mostró flora polimicrobiana epidérmica.
 Masa axilar derecha gigante con datos de necrosis y sobreinfección. B) Lesión tumoral incipiente axilar izquierda.")
La biopsia axilar y ganglionar derecha informaba de un denso infiltrado linfoide atípico (positivo CD3 y CD45, Ki-67: 30-40%) desde hipodermis a epidermis, con marcado epidermotropismo, compatible con linfoma cutáneo primario de células T, tipo MF en estadio tumoral. Se inició tratamiento con prednisona 100mg/día y radioterapia local axilar derecha (16 sesiones) obteniendo remisión en 3 meses. Cinco meses después es hospitalizada por disnea de reposo y deterioro del estado general, destacando en el estudio realizado los hallazgos del TAC toraco-abdominal que mostraba adenopatías múltiples (axilares, mediastínicas, retroperitoneales) que presentaban captación hipermetabólica, así como en bazo, médula ósea, focos cutáneos y pulmonares bilaterales, sugerente de proceso linfoproliferativo. La biopsia ganglionar axilar fue compatible con linfoma no Hodgkin T de alto grado, iniciándose tratamiento quimioterápico (gemcitabina) con ausencia de respuesta y desenlace fatal en el propio ingreso hospitalario.
Los linfomas cutáneos de células T representan un grupo heterogéneo de linfomas no Hogdkin caracterizados por una proliferación clonal de linfocitos T maduros y células natural killer con capacidad de anidamiento cutáneo, siendo el 2% de todos los linfomas y el 75-80% de los linfomas cutáneos primarios1. Presentan una incidencia de 0,5 casos por 100.000 habitantes/año, siendo más frecuente en el sexo femenino en una relación 1.5:12. Entre los LCCT, la MF es el subtipo más frecuente, derivando la célula neoplásica del linfocito TCD4+ y definiéndose histológicamente por la presencia de células T atípicas de núcleo cerebriforme y presencia de infiltrado de epidermis3. La historia natural de la MF se caracteriza por ser tendente a la cronicidad e indolencia, con una fase inicial consistente en placas o máculas en zonas no fotoexpuestas, con posterior progresión en estadios avanzados (10% de los casos) a lesiones tumorales cutáneas y en fases muy avanzadas o tardías puede ocurrir invasión extra-cutánea ganglionar, hematológica o visceral4. No existe tratamiento curativo, presentando diferentes opciones para obtener la remisión, las cuales dependen de la estadificación y situación clínica, entre ellas se encuentran la fototerapia (PUVA), quimioterapia tópica o radioterapia5. La asociación de MF y linfoma en el mismo paciente es excepcional existiendo escasas y puntuales referencias en las que ambas enfermedades se presentan de forma secuencial o simultánea6,7. Diversos estudios relacionan a la MF con un aumento en el riesgo de malignidad y la aparición de segundas neoplasias, especialmente trastornos linfoproliferativos (linfoma de Hodgkin y no Hodgkin) y aunque cronológicamente la enfermedad cutánea puede presentarse antes o después de los trastornos linfoproliferativos, lo habitual es que sea la predecesora, y la aparición del linfoma ocurra años o décadas después8,9. Se ha observado que esta infrecuente transformación cuando ocurre suele ser en estadios avanzados de la enfermedad, especialmente en la fase tumoral, asociando una evolución más agresiva de la enfermedad y supervivencia más corta10. En este caso se conjugan varios factores que lo hacen singular como son el diagnóstico de la MF en fase tumoral presentando una iconografía impactante y, posteriormente, una evolución rápida y fatal a linfoma no Hodgkin, lo cual es excepcional.
