


Introducción
La arteritis de células gigantes es una vasculitis que afecta fundamentalmente a vasos de mediano y gran calibre, con localización más común en las arterias temporales y en las ramas de la aorta1. Aparece en pacientes generalmente mayores de 50 años y, al contrario de lo que sucede en la arteriosclerosis, cuyos síntomas suelen ser lentamente progresivos, en la arteritis de temporal (AT) las manifestaciones clínicas suelen aparecer de forma brusca, y cuando su localización es únicamente extracraneal, el diagnóstico puede resultar sumamente difícil. Su etiología es desconocida, pero diversos estudios demuestran que se trata de una enfermedad desencadenada por algún antígeno no conocido y, aunque no se ha podido aislar ningún agente infeccioso, algunos autores han señalado que determinados gérmenes, como el parvovirus B19 entre otros, pueden tener un papel importante en la patogénesis de la AT2. Además, la aparición de la enfermedad suele seguir un patrón cíclico sugiriendo la hipótesis infecciosa como su causa3. Por otra parte, la existencia de una activación clonal de una pequeña población de linfocitos T CD4+ con una restricción compartida del receptor del linfocito T (RLT) de esa población linfocitaria, apoya también la hipótesis de que las células T expandidas clonalmente son específicas para un determinado antígeno4.
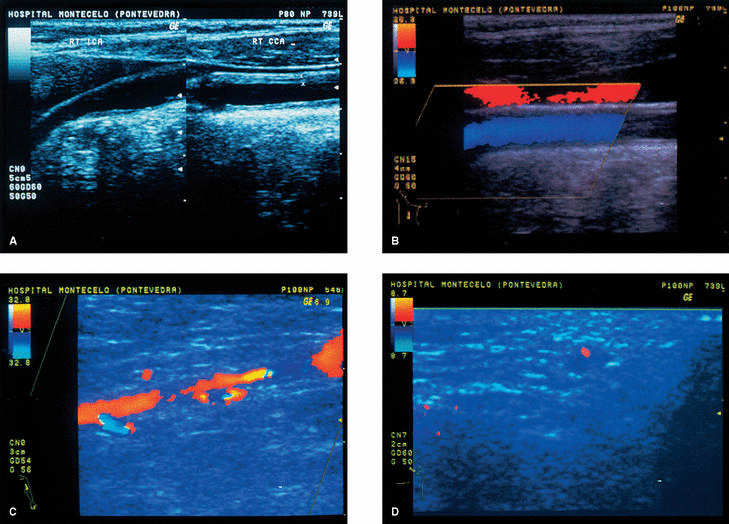
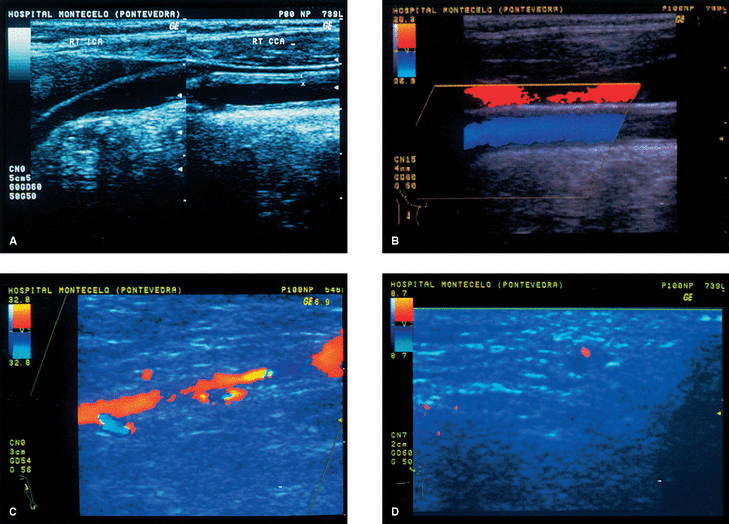
Figura 1. A) Ultrasonidos modo B: engrosamiento circuferencial homogéneo y difuso de la pared de arteria carótida común (entre cursores). B) Ultrasonografía en color de la arteria carótida común sin evidencia hemodinámica de estenosis. C) Ultrasonografía en color de la arteria axilar, que demuestra estenosis de más del 50%, de 2 cm de longitud, con un jet de alta velocidad en la posición distal (color azul). D) Ultrasonografía en color de la arteria temporal. Un plano transversal demuestra un halo hipoecoico alrededor de la luz del vaso (color rojo).
Presentamos un caso de AT con una forma atípica de presentación y sin ninguna sintomatología local craneal que indicase el diagnóstico de la enfermedad. La AT cuando se localiza en miembros, se manifiesta clínicamente como claudicación. Su progresión a isquemia o gangrena es rara, aunque cuando se afectan los grandes vasos se pueden desarrollar aneurismas aórticos, los cuales son una complicación tardía que puede resultar fatal5.
Caso clínico
Mujer de 63 años, sin antecedentes personales de interés, que inició dolor y fenómeno de Raynaud (FR) trifásico de comienzo agudo, de 2 meses de evolución, localizado en la mano izquierda, posteriormente presentó discreta claudicación del miembro superior izquierdo sin pérdida de fuerza. Los únicos hallazgos de laboratorio de interés mostraban una VSG de 47 mm/h, eosinofilia de 10,9% (normal, 1-4%), anticuerpos antinucleares+ de 1/160 y una hematuria microscópica de 15-20 hematíes/campo. En la anamnesis por aparatos no presentó fiebre, cefalea, disfagia ni alteraciones del hábito intestinal, disnea, alteraciones cutáneas, oculares, renales u otras. La paciente tomaba tratamiento con medio comprimido de lorazepam 1 mg al acostarse. El examen físico detectó una ausencia del pulso radial en antebrazo izquierdo, el pulso carotídeo era simétrico y bilateral, con latido normal en ambas arterias temporales. En la auscultación cardíaca y pulmonar no había soplos ni roces. La presión arterial en el brazo derecho era de 160/85 mmHg y en el brazo izquierdo estaba ausente.
Con el diagnostico de fenómeno de Raynaud y con la sospecha de tromboembolia periférica, la paciente se valoró por cirugía vascular sin encontrar patología quirúrgica en relación con el origen del problema, y se prescribió tratamiento con pentoxifilina (400 mg/8 h) por vía oral.
La condición general de la paciente continuó estacionaria durante los meses sucesivos, y se determinó estudio de anticuerpos anticardiolipina IgG e IgM, anti-scl70, anticentrómero, anti-RNP y anti-DNA, con resultados negativos. La radiografía de tórax fue normal. Ante la posibilidad de un origen embolígeno, se realizó un estudio cardiológico, con electrocardiograma y ecocardiograma sin alteraciones.
Con la sospecha posterior de arteritis de grandes vasos, se realizó una eco-Doppler en color de troncos supraaórticos y sus ramas, y se objetivaron cambios estructurales de la pared de las arterias carótida común y axilar izquierda (fig 1A-C). Una angiografía de troncos supraaórticos y de arteria axilar izquierda mostró la existencia de estrechamiento de la luz del vaso de la arteria axilar por engrosamiento de la pared afectada. Con la posibilidad de AT se realizó una eco-Doppler de la arteria temporal izquierda, y se visualizó un halo perivascular compatible con el diagnóstico clínico (fig. 1D). La biopsia de la arteria temporal demostró los hallazgos característicos de la enfermedad.
La paciente se trató con prednisona oral a dosis de 1 mg/kg/día. A los 2 meses de iniciado el tratamiento se realizó una eco-Doppler de control de la arteria temporal afectada, y se observó desaparición total del halo, y en la arteria carótida y sus ramas persistían las lesiones de engrosamiento en arterias carótidas comunes y la estenosis de la arteria axilar y humeral izquierdas. En la arteria temporal izquierda existía interrupción del flujo de sangre en un pequeño trayecto en relación con el engrosamiento de su pared. La presión arterial en el brazo afectado ha permanecido ausente durante los controles posteriores. La paciente prosiguió tratamiento con 0,5 mg/kg/día de prednisona. El tratamiento con esteroides no modificó los cambios estructurales en los grandes troncos después de 5 años.
Discusión
Los hallazgos clínicos de la paciente sugerían un FR de instauración tardía. La forma de presentación, la afección unilateral y la claudicación posterior apuntaban a un proceso vascular periférico de naturaleza obstructiva. Una eco-Doppler de los vasos supraaórticos y sus ramas evidenció alteraciones estructurales de la pared, compatibles con una vasculitis de grandes vasos. Aunque no existían síntomas locales y ante la sospecha de una AT, se realizó también una eco-Doppler de la arteria temporal del lado izquierdo, y se observó la presencia de un halo típico perivascular compatible con la hipótesis clínica. Una biopsia de dicha zona confirmó el diagnóstico.
Se piensa que la AT es una enfermedad causada por un antígeno desconocido que desencadenaría la enfermedad en los pacientes con predisposición genética, ya que existe una preponderancia en la mujer y una asociación con determinados alelos HLA-DRB16. Por otra parte, se le da un papel fundamental a la presencia de macrófagos y linfocitos T en la patogénesis de la enfermedad. Existe una activación de una población de linfocitos T helper CD4+ capaces de reconocer algún antígeno con capacidad para producir interferón gamma (IFN*) el cual actuaría con capacidad para activar los macrófagos que infiltran las paredes de los vasos afectados, con secreción de ciertas citocinas (IL-1, IL-6) y metaloproteinasas que ocasionan inflamación local y la destruccion de la pared del vaso.
La afección de los grandes vasos con síntomas vasculares periféricos se da en un 10-15% de los pacientes con AT7. Los síntomas suelen presentarse como una claudicación o una enfermedad sin pulso. Algunos autores comentan el buen pronóstico en general de esos pacientes con afección de grandes vasos, y ello parece que está relacionado con la expresión de determinadas citocinas in situ en la pared del vaso, así la transcripción in situ de IL-2 es mayor en los pacientes con afección de grandes vasos. Opuestamente, la transcripción in situ de IFN*, especialmente entre los pacientes con AT sin afección vascular periférica, lleva consigo un peor pronóstico con presentación de complicaciones visuales, entre otras. Estos mismos autores sugieren un tratamiento menos agresivo, con esteroides, a los pacientes con AT y afección de grandes vasos como consecuencia de su mejor pronóstico8. En nuestro caso, el tratamiento con esteroides no modificó el curso clínico a pesar de la mejoría clínica experimentada al principio, y por ello debería reconsiderarse su suspensión al cabo de un cierto tiempo.
Como en nuestro caso, es recomendable pensar en la posibilidad de AT ante cualquier paciente mayor de 50 años que presente cualquier fenómeno vascular periférico de origen incierto y no presente datos de enfermedad embolígena. También hay que destacar la presencia de algunos signos clínicos guía, que en algunos casos complementan la sospecha diagnóstica de AT, como son la presencia de una colestasis como resultado de una enfermedad hepática granulomatosa9 o, como en el caso que nos ocupa, la presencia de una discreta hematuria, hallazgo que, como algunos autores han comentado, se presenta al inicio de la enfermedad en un 33% de los casos sin presentar signos de infección urinaria u otra afección concomitante10.
Quisiéramos destacar la importancia del uso de la eco-Doppler en la detección de imágenes de lesión y en la búsqueda de afecciones en las arterias temporales, como apuntan distintos estudios11, y pensamos que su uso se debe hacer extensivo a otras localizaciones como en las ramas que derivan de troncos supraaórticos, sobre todo por su fiabilidad, inocuidad y comodidad en la aplicación de dichas técnicas. Su aplicación sistemática nos ayudaría a la detección precoz, seguimiento y tratamiento de las alteraciones que derivarían, aunque otros autores recomiendan el uso de técnicas más sofisticadas pero más costosas, como la aplicación de resonancia magnética nuclear en la detección de alteraciones estructurales de los vasos en la AT12.