





Introducción
La osteoporosis (OP) causa cada año más de 1,5 millones de fracturas (300.000 de cadera, 700.000 vertebrales y 250.000 de muñeca). Sus costes directos anuales (2001) alcanzaron los 17.000 millones de dólares (47 millones diarios), mientras que los costes indirectos e intangibles (dolor crónico, deterioro de la calidad de vida, etc.) son incalculables y están en permanente aumento1.
El mercado global de tratamientos destinados a frenar el impacto de esta enfermedad se estimó en el año 2001 en 5.500 millones de dólares (incluyendo la terapia hormonal sustitutiva [THS]). La progresión de crecimiento sitúa este mercado en 11.000 millones para el año 2008, de acuerdo con una serie de factores, entre los que destacan el envejecimiento poblacional, un notable y progresivo incremento en los diagnósticos al generalizarse los métodos de detección y la aparición de nuevos conceptos fisiopatológicos con capacidad expansiva de mercado, como la calidad ósea2. Esto explica que, en la actualidad, más de 40 compañías farmacéuticas se encuentren en una fase activa de investigación en el campo de la OP, lo que beneficiará a los pacientes y a la ciencia médica, que verá incrementado su ya amplio contenido conceptual.
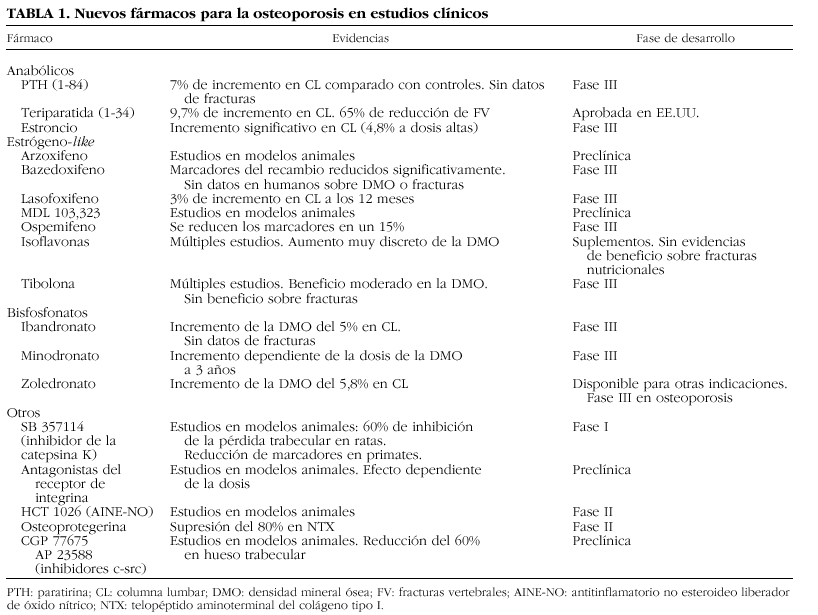
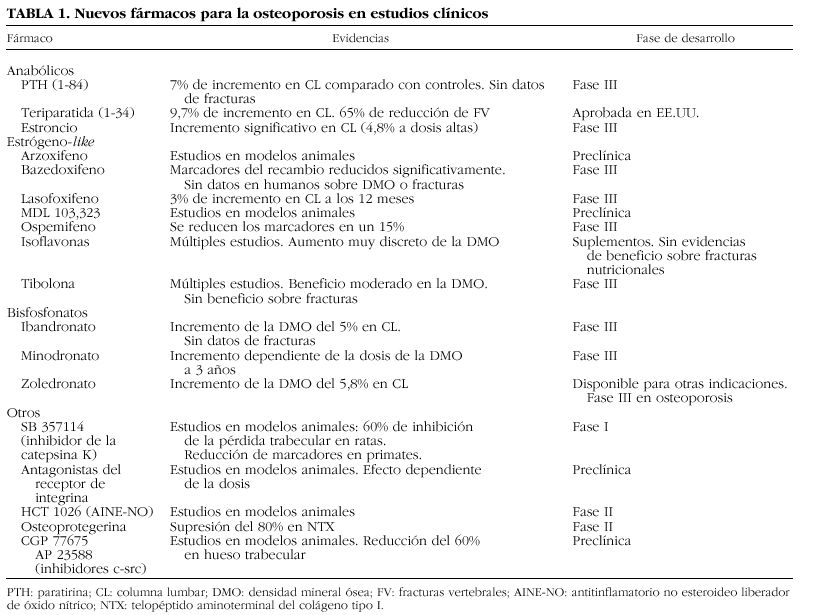
Los avances más recientes en la investigación han mejorado nuestro conocimiento de los mecanismos moleculares que intervienen en la resorción ósea, lo que ha permitido vislumbrar posibles dianas terapéuticas de enorme interés. Aunque las vías que regulan la formación y activación del osteoblasto continúan siendo oscuras y su conocimiento científico es mucho más incipiente, la investigación camina con paso firme también en esta otra orilla del remodelado y ya disponemos de información relevante que puede permitir el desarrollo de nuevas moléculas «osteoformadoras» de potencia desconocida hasta el momento. En la tabla 1 se enumeran algunas de las moléculas más interesantes en fase de desarrollo2. Como se puede observar, las líneas de investigación abarcan desde el desarrollo de familias de fármacos ya conocidas, como los bisfosfonatos o los moduladores selectivos de los receptores estrogénicos (SERM), hasta la aparición de nuevas dianas terapéuticas, lo que ha sido posible gracias a los avances que se han producido en la década pasada en aspectos básicos mal conocidos hasta entonces como la activación y acción de los osteoclastos. En la presente revisión se analizan algunas de las vías de mayor interés, cuya explotación científica podrá permitir una indudable expansión del arsenal terapéutico en los próximos años.
Expansión de las «sagas» farmacológicas actuales
Aunque los fármacos actualmente disponibles tienen un grado de eficacia aceptable, hay varios compuestos experimentales en desarrollo que ofrecen buenas perspectivas a medio plazo (tabla 2).

En el área de la THS los nuevos productos incluyen: progestágenos sintéticos para utilizar en asociación con estrógenos, esteroides sintéticos, tales como el acetato de osaterona; y también los productos naturales como las isoflavonas, ya comercializadas en algunos países, pero cuyas evidencias sobre eficacia y seguridad distan mucho de ser las adecuadas.
Existen una serie de nuevos SERM que podrían mejorar la eficacia de los que ya están comercializados, tanto en el campo de la profilaxis del cáncer de mama dependiente del estrógeno (tamoxifeno) como en la osteoporosis (raloxifeno). Entre los productos de segunda generación que se encuentran en una fase más avanzada de desarrollo, los más prometedores son el bazedoxifeno y el lasofoxifeno. En esta área, no obstante, debemos ser muy cautos ya que, al menos, 3 moléculas han sufrido una suspensión de su desarrollo, a pesar de haber alcanzado la fase 3: droloxifeno (Pfizer), levormeloxifeno (Novo-Nordisk) e idoxifeno (Glaxo). Existen también modelos teóricos de interés como la asociación de determinados SERM con estrógenos, con la intención de potenciar los efectos positivos sobre el hueso y reducir la incidencia de efectos adversos (tabla 3). Por otro lado, tras el descubrimiento del segundo receptor estrogénico3,4, han surgido nuevos intentos para la producción de compuestos que actúen selectivamente sobre uno u otro receptor (véase más adelante).

Los bisfosfonatos (BF), los fármacos actualmente más eficaces para el tratamiento de la OP, son las moléculas cuyo desarrollo mantiene unas expectativas a corto plazo más intensas. Los nuevos BF en desarrollo buscan 2 objetivos fundamentales: mejorar la adherencia (y, por tanto, la eficacia real) y confirmar la seguridad tanto ósea como extraósea. Entre ellos, las 2 moléculas en un estadio de desarrollo más avanzado son el ibandronato y el zoledronato, que proponen formas de administración realmente espectaculares (este último ha mostrado utilidad con inyecciones anuales) aunque deberán aclarar una serie de dudas con estudios a largo plazo.
Las nuevas calcitoninas buscan nuevas formas posológicas que permitan mejorar la adherencia y la tolerabilidad. Por ejemplo, se están desarrollando sistemas de liberación en aerosol y por vía oral (oratonina) y rectal. De forma simultánea, Fida Advanced Biopolymeres está desarrollando un curioso sistema de microesferas de liberación vaginal de calcitonina de salmón, con la intención de conseguir una mejor tolerancia manteniendo una biodisponibilidad similar a la vía inyectable.
La administración intermitente de paratirina (PTH) es el estímulo osteoanabólico más potente de que disponemos. No obstante, su viabilidad comercial continúa suscitando dudas en los ámbitos especializados, que serán resueltas muy pronto pues, en el momento de redactar este manuscrito, estamos a punto de disponer de la primera molécula comercializada, la teriparatida (fragmento recombinante humano 1-34). Aspectos comerciales aparte, la investigación de esta línea está en plena expansión, con la intención de obtener nuevas formas que mejoren la eficacia y seguridad del fármaco. Entre ellas destaca la PTH (1-34) con anillo cíclico entre los residuos 13 y 17, la PTH intacta (1-84) y fórmulas más novedosas de liberación intranasal e inhalada.
Por último, debemos destacar 2 líneas del ranelato de estroncio, fármaco en desarrollo clínico avanzado que ha mostrado un aumento de la densidad mineral ósea (DMO) dependiente de la dosis y que actúa principalmente como antirresortivo, aunque también se le atribuyen propiedades anabólicas. No podemos decir nada más por el momento.
El futuro del tratamiento antirresortivo
El osteoclasto (OC) es la célula que se encarga de degradar y extraer la matriz calcificada, para lo que contiene una serie de estructuras especializadas, con altos requerimientos metabólicos, que han sido bien caracterizadas en los últimos años y que han permitido avanzar en el conocimiento de dianas terapéuticas de indudable interés. El proceso de formación del OC está modulado por diferentes hormonas y citocinas. Tras la maduración, y ya en forma de célula multinucleada, migra a la superficie quiescente, donde se ancla firmemente mediante la interacción de la integrina alfa-V beta-3 (receptor de vitronectina) con algunas proteínas de la matriz, como la osteopontina, formando una zona de sellado que puede ser considerada un lisosoma extracelular. Dentro de este espacio, son liberados protones que mantendrán un pH muy bajo que consigue disolver el mineral y liberar proteínas de matriz que son, finalmente, degradadas por la catepsina K. Disponemos, por tanto, de múltiples lugares o dianas terapéuticas, que pueden ser bloqueadas para interrumpir la resorción ósea (tabla 4).

Antagonistas de la señal RANK-RANKL
En 1990 se estableció que la transformación in vitro de macrófagos en OC requería la presencia de células del estroma o de su progenie osteoblástica5. Tras una década de confusión, está plenamente establecido que estas células accesorias expresan las 2 moléculas que son esenciales y suficientes para promover la osteoclastogénesis6, el factor estimulador de las colonias macrofágicas (M-CSF) y el ligando del receptor para la activación del factor nuclear kappa B (RANKL), también conocido por otros nombres, ya en desuso, como OPGL y TRANCE. El M-CSF interviene en las fases iniciales de la maduración macrofágica, a través de su receptor, c-Fms, proporcionando, de esta forma, las señales adecuadas para la supervivencia y proliferación de los futuros osteoclastos. Cuando los osteoblastos (OB) o sus precursores estromales reciben señales osteoclastogénicas (PTH, estímulos mecánicos, etc.) sobreexpresan RANKL y M-CSF y frenan la producción de osteoprotegerina (OPG). Estas 2 moléculas interaccionan con sus receptores en las células monocitomacrofágicas, e inducen su diferenciación en OC, un proceso que la OPG inhibe. El OC diferenciado se polariza hacia la superficie ósea, mediante señales derivadas de la integrina alfa-V beta-3, y se produce su fuerte anclaje mediante una zona de sellado que deja en su interior un hueco en el que se va a producir la resorción. A continuación, mediante señales que activan el citoesqueleto celular, se produce el ribete en cepillo y la acidificación del medio extracelular mediante una bomba de protones, manteniendo el pH intracelular por intercambio HCO3/Cl. El medio ácido moviliza la fase mineral del hueso y proporciona un microambiente óptimo para la acción degradante de la matriz, llevada a cabo por la catepsina K.
La OPG es una glucoproteína de la superfamilia del receptor del factor de necrosis tumoral (TNFR), que carece de la secuencia hidrofóbica de membrana y se comporta como antagonista endógeno de la vía de transducción de la señal RANK/RANKL. Se encuentra en fase de desarrollo avanzado en osteoporosis7, cáncer8,9 y artritis10. Estudios preliminares han mostrado que la administración parenteral de OPG provoca una profunda inhibición de la resorción, considerablemente más poderosa que la que se consigue con los BF disponibles. Aún es prematuro asumir que este fármaco pueda proporcionar un óptimo control del remodelado sin afectar a la resistencia ósea y sin provocar efectos adversos extraóseos. No obstante, la acción sobre el sistema RANKL/OPG puede considerarse como un nuevo ámbito terapéutico de indudable interés, con enormes expectativas, que han permitido el diseño de líneas de investigación muy variadas, que van desde la inmunoterapia11,12 con autovacunas RANKL (inmunización activa con proteínas RANKL modificadas que contienen péptidos extraños inmunodominantes, fácilmente reconocibles por los linfocitos T-helper, con la intención de inducir la producción de Ac neutralizantes por estas células) hasta la identificación de moléculas pequeñas que aumenten la actividad del promotor de OPG y supriman la diferenciación de OC. En este sentido, se están investigando varios fármacos (LY 481736, 541727, 481736) que, en modelos preclínicos, han mostrado que inhiben la resorción in vitro, previenen la hipercalcemia en ratas paratiroidectomizadas tratadas con PTH, reducen el número de lesiones y su área en ratas con metástasis óseas y la resorción ósea en ratas con artritis por adyuvante.
Antagonistas de la integrina alfa-V beta-3
Esta integrina parece ser el mediador clave de la unión del OC a la matriz ósea y, por tanto, de la formación de la zona de sellado, imprescindible para que se produzca una adecuada resorción13. Debido a que todos sus ligandos (osteopontina, fibronectina, etc.) contienen la secuencia peptídica Arg-Gly-Asp (RGD), se están desarrollando antagonistas que contengan estas secuencias RGD para inhibir la unión del OC a la matriz. Además, el bloqueo mediante anticuerpos monoclonales provoca una fuerte inhibición de la resorción in vivo14, lo que animó a varios grupos a desarrollar líneas de investigación que analizaran las características diferenciales de esta molécula para diseñar fármacos específicos que eviten los potenciales efectos adversos extraóseos. El SB 265123 es el prototipo en fase más avanzada de desarrollo y ha mostrado una gran capacidad antirresortiva en ratas tiroparatiroidectomizadas (frenaba la reabsorción inducida por PTH en más del 80%) y en ratas ovariectomizadas, tanto por vía oral como intravenosa y de manera dependiente de la dosis15.
Inhibidores de la catepsina K
La catepsina K (EC 3.4.22.38) es una proteasa cisteína alta y selectivamente expresada por los OC anclados en la superficie de reabsorción, que facilita la degradación de la matriz. La importancia de esta proteasa para la resorción adecuada se demostró en ratones transgénicos y en la rara enfermedad llamada picnodisostosis (enfermedad de Toulouse-Lautrec), en la que una mutación provoca la eliminación funcional de esta enzima y un fenotipo osteoesclerótico y de talla baja. El SB 331750 es un inhibidor de la catepsina K que ha mostrado eficacia antirresortiva en tipos de rata y en primates no humanos.
Inhibidores c-src
La cinasa c-rsc es una proteína tirosina asociada a membrana sin características de receptor, que pertenece a una familia de 8 miembros relacionados. Su papel en la reabsorción es sobradamente conocido y, recientemente, se ha especulado acerca de su papel en la diferenciación osteoblástica. Se están desarrollando inhibidores de c-rsc que, además de su capacidad antirresortiva, podrían tener capacidad anabólica. El llamado PP 2 es el prototipo de esta nueva clase de fármacos y, en algunos tipos de ratones, ha mostrado su capacidad terapéutica in vivo, lo que ha impulsado su experimentación en modelos más avanzados, aunque aún no ha entrado en fase clínica.
Acciones estrogénicas: un potencial con futuro
Los estrógenos tienen múltiples acciones en diversos órganos diana. En el esqueleto son potentes inhibidores de la resorción, lo que explica que tras la menopausia se acelere la renovación y se produzca un balance negativo, que contribuye de manera decisiva a la producción de la osteoporosis. Aunque los mecanismos por los que se produce esta acción positiva de los estrógenos sobre el esqueleto son poco conocidos, se sabe que inhiben la producción de interleucina y del M-CSF por las células del estroma medular, incrementando su producción de OPG, con lo que frenan la maduración y activación de los OC. El papel de los estrógenos en el tratamiento de la osteoporosis ha quedado plenamente establecido en estudios observacionales y en ensayos clínicos controlados. Sin embargo, su utilización a largo plazo provoca una serie de efectos adversos potenciales, lo que ha provocado un cierto rechazo a su prescripción en enfermedades crónicas como la OP. Por ello, se han intensificado los esfuerzos para conocer los mecanismos moleculares de las acciones estrogénicas que participan en la selectividad molecular, para diseñar fármacos, que careciendo de las acciones deletéreas de estas hormonas, mantengan o incrementen su potencial preventivo de fracturas. Los SERM son las primeras moléculas que presentan selectividad, aunque la acción del raloxifeno (único aprobado en la OP) sobre el tejido óseo no parece muy potente y solamente ha mostrado beneficios en fracturas vertebrales. Nuevos SERM, cuyo potencial estamos a la espera de comprobar, están en fases muy avanzadas de desarrollo. Pero se continúan explorando las acciones estrogénicas para conseguir nuevos fármacos que mejoren su potencial y que sean diferentes de los SERM. Para analizar estas vías es preciso recordar algunos nuevos hallazgos.
Hay dos receptores estrogénicos, alfa y beta, que son moléculas proteicas de 595 y 485 aminoácidos, respectivamente, y que se comportan como factores de transcripción regulados por el ligando. Presentan diferentes patrones de distribución tisular y pueden ser activados con diferentes afinidades por compuestos esteroideos y no esteroideos. Cuando la hormona se une al receptor hay un cambio estructural que permite a este complejo unirse a secuencias específicas del ADN que ponen en marcha el aparato transcripcional. La capacidad del ligando para afectar a la estructura tridimensional de la región AF-2 del receptor es un importante mecanismo que contribuye a la diferente selectividad de algunos fármacos. En particular, se ha observado, en estudios estructurales, que la orientación de la hélice de transactivación C terminal (H12) es sensible a la naturaleza del ligando, y se orienta hacia el interior de la cavidad en presencia de agonistas, que son introducidos completamente en la cavidad que forma el dominio de unión al ligando. Sin embargo, en presencia de un antagonista, cambia de orientación impidiendo la entrada completa de la molécula, lo que proporciona un modelo estructural para explicar algunas respuestas específicas16.
Acciones no genotrópicas de las hormonas sexuales
Este término se aplica a un amplio espectro de efectos de las hormonas sexuales, que varían desde cambios fisicoquímicos a acciones independientes del receptor, incluyendo además la activación de posibles receptores de membrana. Se han observado potentes acciones antiapoptóticas en OB y OC murinos, que se producen a través de cascadas de señal, vía acción no genotrópica. A diferencia de las acciones clásicas de los esteroides en los tejidos reproductivos, éstas no son específicas del sexo y requieren sólo el dominio de unión al ligando. Este concepto ha permitido el desarrollo de un grupo de moléculas, denominadas con el acrónimo ANGELS (activator of nongenotropic estrogen-like signaling) que se caracterizan por su reducido tamaño y porque aunque simulan las acciones no genotrópicas de los estrógenos, carecen de sus efectos sobre los receptores nucleares clásicos (acciones genotrópicas). Un producto sintético llamado Estreno®, es el prototipo de esta línea de fármacos potenciales. Carece de la actividad transcrip-
cional clásica, pero reproduce todas las acciones no genotrópicas de los esteroides sexuales17-20.
Estimuladores de la osteoformación
La formación ósea está regulada por factores de crecimiento expresados en las células óseas, que son incorporados a la matriz y liberados activamente cuando el hueso es reabsorbido. Estos factores estimulan la proliferación y diferenciación de los OB, que preceden a la síntesis de la matriz mineralizada20-25. Aunque los factores que controlan la línea osteoblástica y, en definitiva, la osteoformación son mucho menos conocidos, existe un notable interés por conocer posibles dianas terapéuticas que permitan estimular la formación y superar ese «techo terapéutico» que caracteriza, al menos desde el punto de vista teórico, a los antirresortivos.
Son pocas las moléculas en desarrollo y, dada la brevedad de esta revisión, no podemos extendernos en el análisis de este campo en profundidad. Solamente destacaremos algunas de las moléculas con mayor potencial, como las proteínas morfogenéticas del hueso (BMP) y sus análogos.
Proteínas morfogenéticas del hueso
Fueron descubiertas en 1965 por Marshall R. Urist, quien observó que la implantación intramuscular de matriz ósea desmineralizada desencadenaba la formación de hueso26. Las BMP son factores de diferenciación pleiotrópicos que regulan el destino celular dirigiendo una serie de actividades transductoras en cascada. Pertenecen a la superfamilia del TGF ß27,28 (excepto la BMP-1, de la familia de las metaloproteasas) y actúan a través de receptores heterodiméricos con actividad serina-treonina cinasa29,30.
Las BMP pueden inducir la diferenciación de células mesenquimales en osteoblastos y simultáneamente inhibir su diferenciación en otras estirpes celulares, cambiando la expresión genética de estas células31. La angiogénesis es también un factor crítico en la neoformación ósea, y se ha observado que las BMP inducen expresión de péptidos angiogénicos en las células mesenquimales que actúan de forma paracrina sobre las células endoteliales31. El papel que estas poderosas moléculas desempeñan en la clínica es muy prometedor, pero sus acciones positivas sobre la diferenciación osteoblástica sólo han podido ser utilizadas localmente en fracturas o cirugía de raquis, donde aceleran la reparación de las fracturas de manera espectacular. La BMP-2 ha sido la más empleada y es el factor osteogénico más potente que se conoce, lo que provoca que cuando se utiliza por vía intramuscular o intravenosa desencadene una rápida calcificación muscular o vascular, respectivamente. Por ello, se están investigando nuevos fármacos que, aunque carezcan de estas acciones directas de las BMP, tengan capacidad estimuladora de la actividad osteoblástica y puedan resultar útiles en la osteoporosis.
La AC 100 es un péptido de 22 aminoácidos que forma el núcleo central de MEPE (fosfoglucoproteína de la matriz extracelular, expresada en osteoblastos normales). Es equipotente a la BMP-2 con respecto al estímulo de la proliferación de OB, pero no provoca efectos adversos locales, y se encuentra en fase de desarrollo preclínico (estudios en fase I en humanos32). Los fragmentos de la DBP (vitamin D binding protein) son péptidos de 3 a 14 aminoácidos formados por reducción simple a partir de un péptido original de 14 aminoácidos del tercer dominio de la DBP humana, que presentan una notable acción estimuladora de la formación, y se encuentran en fases muy iniciales de desarrollo, pero con un futuro prometedor33.
Por último, otro factor anabólico que se está experimentando en modelos animales es el factor de crecimiento fibroblástico tipo 2 (FGF-2), con una potente acción anabólica por vía intravenosa, aunque con acciones extraóseas que limitan sus posibilidades de utilización en la OP.
Direcciones futuras
Hasta el momento se han descubierto más de 60 proteínas asociadas con vías reguladoras del osteoclasto, aproximadamente 10 veces el número identificado hace 3 años. Algunas de ellas son excelentes dianas terapéuticas, por lo que es lógico suponer que, a corto plazo, los nuevos fármacos tengan una acción antirresortiva.
Sin embargo, el conocimiento de la biología del osteoblasto está comenzando a situarse en cotas impensables hace unos años. Hay que destacar el enorme impacto que ha causado en la cirugía ortopédica la irrupción de las BMP. Por ello, no es utópico pensar que, a medio plazo, estarán disponibles fármacos con acción osteoformadora espectacular que conseguirán superar el «techo terapéutico» de los antirresortivos.
El futuro de la investigación farmacológica dependerá, siguiendo a Collins33, de la confluencia de los 3 pilares fundamentales que soportan la investigación actual en esta área: desarrollo del proyecto Genoma Humano (nuevas dianas terapéuticas), avances en química combinatoria (nuevas moléculas) y crecimiento de la tecnología robótica (capacidad de cribado molecular). Nos encontramos, por tanto, en una fase inicial del tratamiento de la os teoporosis, y los avances aquí resumidos nos deben servir de estímulo para tratar a nuestros pacientes con los fármacos disponibles de la forma más eficaz posible, para conseguir que sus huesos lleguen en buenas condiciones a ese futuro cuyo horizonte comienza a vislumbrarse cada vez más despejado.