





A multicenter study was conducted. A panel containing DNA from Histoplasma capsulatum, as well as negative and cross-reaction controls, was sent to five different laboratories, members of the MICOMOL network from CYTED Program.
AimsThe objective was to assess the accuracy of different PCR protocols to detect H. capsulatum DNA.
MethodsSeven different PCR protocols were tested. They were based on PCR techniques and used unicopy and multicopy targets.
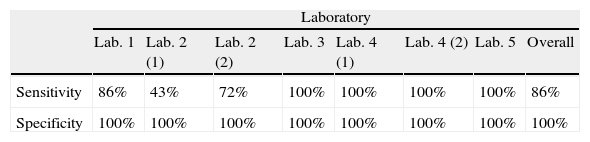
ResultsMost of these protocols (4/7) were able to detect the smallest amounts of fungal DNA (102fg/μl). Overall sensitivity was 86% and specificity was 100%. The protocol based on a unicopy target (SCAR220) presented lower sensitivity (43%) but 100% specificity. The real-time protocols tested were highly reproducible, sensitive, and specific. Neither false positives nor cross-reactions were detected in any protocol.
ConclusionsAll laboratories were able to amplify H. capsulatum DNA, and real-time PCR seems to be a promising tool to efficiently detect this pathogen in clinical samples.
Se realizó un estudio multicéntrico en el que participaron cinco laboratorios miembros de la red MICOMOL a partir del programa CYTED. Los participantes recibieron un panel que contenía muestras de ADN de Histoplasma capsulatum, al igual que paneles con muestras de control negativas y de reacciones cruzadas.
Objetivosel objetivo del presente estudio fue examinar la precisión de los diferentes protocolos de PCR para la detección de ADN de H. capsulatum.
MétodosSe examinaron siete protocolos, todos ellos basados en técnicas de PCR, que empleaban como diana plásmidos unicopia o multicopia.
Resultadosla mayoría de estos protocolos (4/7) pudieron detectar las cantidades más pequeñas de ADN (102fg/μl). La sensibilidad global fue del 86% y la especificidad del 100%. El protocolo basado en el plásmido unicopia SCAR220 se asoció a la menor sensibilidad (43%) pero a una especificidad del 100%. Los protocolos de PCR en tiempo real fueron muy reproducibles, sensibles y específicos. En ningún caso se detectaron resultados falsos positivos ni reacciones cruzadas.
Conclusionestodos los laboratorios pudieron amplificar el ADN de H. capsulatum y las técnicas basadas en PCR en tiempo real parecen un instrumento prometedor para la detección eficiente de este patógeno en muestras clínicas.
Histoplasmosis is an infection acquired by the inhalation of Histoplasma capsulatum conidia present in soils rich in organic matter, particularly guano of bats or birds.8 Although this fungus has a global distribution, the most endemic regions are found in the American continent: Mississippi, Ohio, and Missouri river valleys in United States of America, and Central and South America.6,7 Nowadays, it is the most common endemic mycoses in Europe, mainly due to immigration and travellers coming from these endemic areas.2
Microbiological classic diagnosis is based on isolating the organism in cultures, examining microscopically fluids and tissues, and doing serological techniques.12 Nonetheless, all these methods have limitations. Cultures are slow, can take up to 3–4 weeks to be positive, and require level 3 facilities; the sensitivity and specificity of histopathological and serological techniques are quite different depending on the patient's clinical condition, thus the histopathology has a reduced sensitivity in the subacute or chronic forms of the pulmonary histoplasmosis, and serological tests are less sensitive in the progressive disseminated forms, probably because of underliying immunosupression. In the acute forms, serology may be negative during the first 2 months of infection. Furthermore, a positive serology result can be misleading in patients from endemic regions because serologic test remain positive for several years.21 These limitations have led to the development of molecular techniques. In the last few years, PCR assays have been proposed for detection of H. capsulatum. Several conventional assays to detect H. capsulatum DNA, targeting single or multicopy genes, have been reported.3,7,10,18,20 Real-time PCR techniques showing a high sensitivity and specificity and yielding results in a few hours4,11,14 have also been published. All these molecular methods are faster than cultures and avoid the manipulation of the fungus, but although these techniques seem very promising, all of them are in-house methods with limited availability and without external validation of results.
In the present study, we performed an interlaboratory comparison of PCR protocols to detect H. capsulatum DNA. The objective was to evaluate the protocols routinely used in each laboratory using a common source of fungal DNA. The laboratories implicated were part of the MICOMOL-Network that emerged from the CYTED programme (Ciencia y Tecnología para el Desarrollo) which is an intergovernmental programme of multilateral cooperation in Science and Technology that aims at cooperation in research and innovation for the development of the Iberoamerican region.
MethodsParticipantsFive laboratories were involved in this study. Four of them are located in endemic countries (Argentina, Brazil, Colombia, and México) and one in a non-endemic region (Spain); all are members of the MICOMOL-CYTED network. The centers have both clinical and scientific expertise in the field and use PCR techniques in routine laboratory tasks.15,20 They have been designated with the following numerical code: (Lab. 1) Unidad de Micología Médica y Experimental, Corporación para Investigaciones Biológicas, Medellín, Colombia; (Lab. 2) Departamento de Microbiología y Parasitología, UNAM, México D.F., Mexico; (Lab. 3) Laboratório de Micologia, Instituto de Pesquisa Clinica Evandro Chagas, Fundação Osvaldo Cruz, Rio de Janeiro, Brazil; (Lab. 4) Servicio Micosis Profundas, Departamento de Micología, INEI-ANLIS Dr. Carlos G. Malbrán, Buenos Aires, Argentina; and (Lab. 5) Servicio de Micología, Instituto de Salud Carlos III, Madrid, Spain.
Two of these laboratories (Lab. 2 and Lab. 4) contributed with two different protocols and were designated as Lab. 2(1), Lab. 2(2), and Lab. 4(1), Lab. 4(2), respectively. The rest of the centers contributed with a single protocol.
Panel of DNAsA blind panel with ten tubes containing different concentrations of fungal DNA was sent to each participant laboratory in May 2011 (see Table 1). Tubes with different concentrations of H. capsulatum DNA were included together with tubes containing water as negative controls and a tube with Paracoccidioides brasiliensis DNA. To avoid DNA degradation in freeze–thaw, aliquots containing samples were frozen and sent to the laboratories. Each laboratory thawed the panel samples only to perform the assays. To validate the panels, initial testing was performed by the organizing group before their distribution. The receiving centers were asked to confirm the panel reception, comment on its state and keep the tubes frozen at −20°C until tested.
StrainsClinical strains of H. capsulatum (CNM-CM-2721) and P. brasiliensis (CNM-CM-2908) belonging to the microorganism collection of the Spanish National Centre of Microbiology were used to extract DNA.
Each participant center used their own strains to include as positive controls in their respective PCR assays.
DNA extractionDNA extraction from the strains was performed in biosafety level III facilities in compliance with Spanish law.1 Briefly, the strains were grown statically at 30°C in 10ml of GYEP broth (2% glucose, 0.3% yeast extract, 1% peptone) for a week. Mycelia were harvested, dried and introduced in a Falcon tube with six glass beads of 4mm diameter (Sigma–Aldrich Química, Madrid, Spain). The mycelia were subjected to two cycles of freezing in liquid nitrogen for 30s and vortex for 30s to obtain a fine powder. Then, the kit “DNeasy Plant Mini Kit” (Qiagen, Izasa, Madrid, Spain) was employed to extract DNA from the obtained powder following the manufacturer's instructions. The quality and amount of DNA obtained was checked spectrophotometrically.
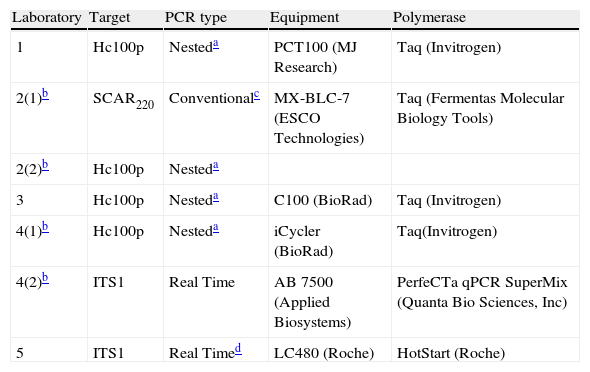
PCR protocolsThe participants were told to use their current amplification systems. A total of seven protocols were tested. These protocols were based on conventional, nested and real time PCR and targeted different DNA regions. The enzymes, the target and the equipment used by each laboratory are described in Table 2. Reactions were performed in a final volume of 20μl, and 2μl of DNA from each tube in the panel were added to the reaction, with the final DNA concentration in the PCR tube being tenfold lower than in the original tube. Each laboratory included positive and negative controls.
Assay details of PCR and equipment used by the five participant teams.
| Laboratory | Target | PCR type | Equipment | Polymerase |
| 1 | Hc100p | Nesteda | PCT100 (MJ Research) | Taq (Invitrogen) |
| 2(1)b | SCAR220 | Conventionalc | MX-BLC-7 (ESCO Technologies) | Taq (Fermentas Molecular Biology Tools) |
| 2(2)b | Hc100p | Nesteda | ||
| 3 | Hc100p | Nesteda | C100 (BioRad) | Taq (Invitrogen) |
| 4(1)b | Hc100p | Nesteda | iCycler (BioRad) | Taq(Invitrogen) |
| 4(2)b | ITS1 | Real Time | AB 7500 (Applied Biosystems) | PerfeCTa qPCR SuperMix (Quanta Bio Sciences, Inc) |
| 5 | ITS1 | Real Timed | LC480 (Roche) | HotStart (Roche) |
Lab. 2 tested the SCAR220 marker (Sequence-Characterized Amplified Region) specific for H. capsulatum. In this protocol, a 220bp fragment was amplified9 using the primers 1281-1283220F (5′-CATTGTTGGAGGAACCTGCT-3′) and 1281-1283220R (5′-GAGCTGCAGGATGTTTGTTG-3′) in the following conditions: denaturation at 94°C for 3min and then 30 cycles of 94°C for 30s, 55°C for 30s; 72°C for 2min and a final extension step at 72°C for 5min. PCR products were subjected to electrophoresis in agarose gels following the protocols of Sambrook and Russel19 to confirm the PCR results. The amplicons were purified using the QIAQuick purification Kit (Qiagen, Valencia, CA) and sequencing was performed using ABI equipment (Applied Biosystems Inc., Foster City, CA).
Protocols based on 100-kDa-like proteinLabs. 1, 2, 3 and 4 used the method described by Bialek et al.3 that targeted the 100-kDa-like protein unique in H. capsulatum. This protein has been described as essential for the survival of the pathogen.16 This protocol was a nested conventional PCR assay in which the first round amplified a 391bp fragment and the second round a 210bp fragment. Primers and PCR conditions were as described by Bialek et al.3 PCR results were verified by using electrophoresis in agarose gels.19
Protocols based on Real Time PCR (ITS1 region)Lab. 4 used a real-time PCR assay that targeted the multicopy ITS1 region of the ribosomal DNA and used the specific primers HcITS-54F (5′-ACCCTTGTCTACCGGACCTGTT-3′ and HcITS-204R (5′-TTTTGACTGGATTATTATCGCTCTCA-3′) and the Taqman probe HcITS-155 (5′ FAM-CGGTGAACGATTGGCGTCTGAGC-TAMRA 3′). These primers and probes were designed using the Program Primer Express® Software version 3.0 (Applied Biosystems). Each reaction mixture contained 0.5μM of each primer and 0.2μM of probe. The cycling conditions included the next steps: 2min at 50°C, 10min at 95°C and then 50 cycles as follows: 15s at 95°C and 1min at 60°C. Lab. 5 used a previously described real-time PCR assay4,5 that had been validated in clinical samples of patients with imported histoplamosis.5 This protocol used specific FRET probes that targeted the ITS1 region of ribosomal DNA and included an internal control. For both assays, standard curves were constructed with PCR results from five repetitions of different dilutions of genomic DNA of H. capsulatum. Dilutions ranged between 107 and 1fg DNA/μl. A line (y=mx+b) was constructed by plotting the standard curve of log quantity versus its corresponding CT value, a cycle in which fluorescence becomes detectable from the background. An average value and a 99% confidence interval for crossing-point values obtained for each DNA concentration were calculated.
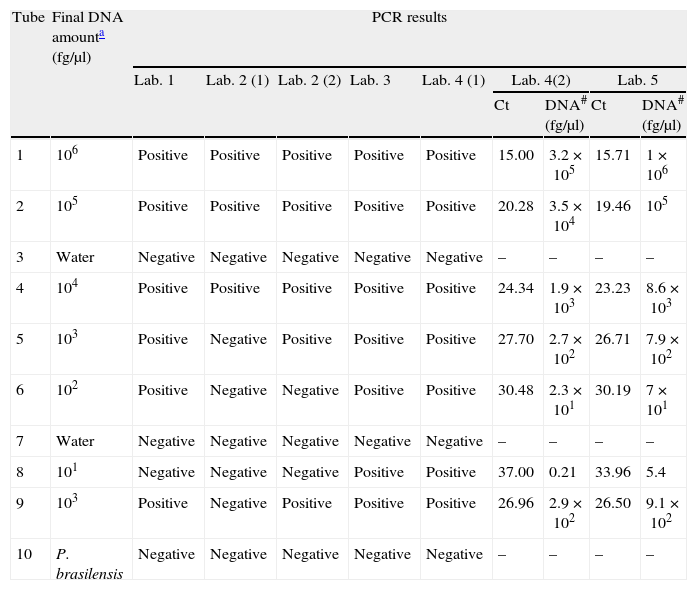
ResultsParticipants submitted results at the end of September 2011. All centers were able to amplify H. capsulatum DNA and 100% of protocols detected the highest DNA concentrations (Tubes 1, 2, and 4). Results are summarized in Table 3. Sensitivity and specificity values for each protocol are shown in Table 4.
PCR results for the seven PCR protocols tested.
| Tube | Final DNA amounta (fg/μl) | PCR results | ||||||||
| Lab. 1 | Lab. 2 (1) | Lab. 2 (2) | Lab. 3 | Lab. 4 (1) | Lab. 4(2) | Lab. 5 | ||||
| Ct | DNA# (fg/μl) | Ct | DNA# (fg/μl) | |||||||
| 1 | 106 | Positive | Positive | Positive | Positive | Positive | 15.00 | 3.2×105 | 15.71 | 1×106 |
| 2 | 105 | Positive | Positive | Positive | Positive | Positive | 20.28 | 3.5×104 | 19.46 | 105 |
| 3 | Water | Negative | Negative | Negative | Negative | Negative | – | – | – | – |
| 4 | 104 | Positive | Positive | Positive | Positive | Positive | 24.34 | 1.9×103 | 23.23 | 8.6×103 |
| 5 | 103 | Positive | Negative | Positive | Positive | Positive | 27.70 | 2.7×102 | 26.71 | 7.9×102 |
| 6 | 102 | Positive | Negative | Negative | Positive | Positive | 30.48 | 2.3×101 | 30.19 | 7×101 |
| 7 | Water | Negative | Negative | Negative | Negative | Negative | – | – | – | – |
| 8 | 101 | Negative | Negative | Negative | Positive | Positive | 37.00 | 0.21 | 33.96 | 5.4 |
| 9 | 103 | Positive | Negative | Positive | Positive | Positive | 26.96 | 2.9×102 | 26.50 | 9.1×102 |
| 10 | P. brasilensis | Negative | Negative | Negative | Negative | Negative | – | – | – | – |
Ct: threshold cycle; #: amount of DNA estimated from the standard curve in the reaction tube.
The protocol based on SCAR marker [Lab. 2(1)] presented the lowest sensitivity for the panel sent (43%) and was not able to detect DNA concentrations of less than 104fg/μl. However, this protocol did not produce false positives or cross-reactions.
PCR results for 100-kDa-like protein protocolsThe limit of detection was different for the four laboratories that performed this protocol. In Lab. 3 and Lab. 4 the protocols based on the 100-kDa-like protein were able to detect the different H. capsulatum DNA concentrations used, reaching the lowest concentration (10fg/μl). In Lab. 1 and Lab. 2(2), fungal DNA concentrations in the PCR reaction tube of less than 102 and 103fg/μl, respectively, were not detected. The overall sensitivity for the panel using this protocol was 89.5%. The detection limit was established between 103 and 10fg/μl. False positives were not detected in any case.
PCR results for protocols based on real-time PCRBoth protocols based on real-time PCR were able to detect the lowest concentrations of DNA (10fg/μl).
When DNA concentration was estimated based on the calibration curves, the average of relative errors was 77% for Lab. 4 and 18.5% for Lab. 5 (Table 3).
Neither false positives nor cross-reactions were detected with these protocols.
DiscussionThis is the first multicenter study addressed to evaluate molecular protocols used to detect H. capsulatum DNA. A blind panel with ten tubes was sent to five different laboratories with the main purpose of assessing the analytical sensitivity of seven PCR protocols to detect H. capsulatum DNA. The results presented in this study show that all laboratories were able to amplify H. capsulatum DNA. All the protocols were specific and no false positive results were detected although some of the PCR schemes used were based on nested PCR which is more prone to produce false positive results. The overall sensitivity and specificity for the panel was 86% and 100%, respectively. Regarding sensitivity, the protocol based on SCAR marker9 presented a sensitivity of 43%, while the nested PCR protocols based on amplification of the gene coding for the 100-kDa-like protein3 presented a high sensitivity. However, Lab. 1 was unable to detect the lowest concentration (10fg/μl) and Lab. 2 only detected up to 103fg/μl of fungal DNA; the other two laboratories (3 and 4) detected all tested fungal DNA concentrations. Consequently, the overall sensitivity for these protocols was 89.5%. The differences in the limit of detection obtained between laboratories that used the same protocol (100-kDa-like protein) may be due to the different PCR equipment, the polymerase or other technical aspects. Further studies will be conducted. Finally, protocols based on real-time PCR detected the lower concentrations and presented 100% sensitivity and specificity.
As we expected, there was a correlation between sensitivity and the PCR target. The unicopy target as SCAR marker exhibited a lower sensitivity than protocols based on multicopy target (ITS1) or nested PCR assays. Nested PCR has the problem of high risk of contamination,23 although in this study no laboratory produced false positive results, which reflects appropriate precautions to avoid DNA-contamination. Real-time protocols were the most accurate and results were generated in a short time. In addition, no further post-PCR analysis such as electrophoresis or sequencing was required. Regarding the quantification of DNA in real-time protocols, there were important differences between Lab. 4 and Lab. 5, suggesting differences in the quantification of the DNA that was used as template to construct the standard curves; a further study is needed to improve this pitfall. In recent years, there have been several multicenter studies with the aim of reaching a consensus on PCR techniques, especially for the detection of fungal genera such as Aspergillus or Candida.17,22,24 In these works, spiked samples with known quantities of a microorganism were distributed among the different laboratories. The methodologies and platforms were used according to the requirements of each laboratory. Some of these works are the result of the recently created working group “European Aspergillus PCR Initiative” (EAPCRI) which aims to standardize molecular methods to detect Aspergillus fumigatus in blood and serum. Different reports with the results of multicenter collaborative studies have been published.13,25 Recommendations for DNA extraction, sample volumes and PCR protocols have been forwarded based on the results obtained from these multicenter assays.
In this collaborative work, we can conclude that the protocols based on multicopy targets are more efficient in the detection of the smallest amounts of DNA, making them more suitable for their use in diagnosis. Moreover, real time PCR protocols were highly sensitive, and specific and they seems to be a promising tool to efficiently detect this pathogen in clinical samples. This work is the first joint effort to achieve a consensus on the PCR methods for the detection of H. capsulatum DNA. Although clinical samples were not used, this study allowed assessing the molecular techniques used by laboratories that diagnose histoplasmosis routinely. Future collaboration will be established to determine the best PCR protocol and the clinical samples to be used for the molecular diagnosis of histoplasmosis.
Conflict of interestThe authors declare that there is no conflict of interest.
The authors thank to the coordinator of the MICOMOL-CYTED network, Dra. Luz Elena Cano and the rest of the members; Corporación para Investigaciones Biológicas (Medellín, Colombia); Escuela de Microbiología (Universidad de Antioquia, Medellín, Colombia); Instituto de Pesquisa Clínica Evandro Chagas (Fundação Oswaldo Cruz, Rio de Janeiro, Brazil); Servicio de Micología, (Centro Nacional de Microbiología, Instituto de Salud Carlos III, Madrid, Spain); Departamento de Micología del INEI-ANLI (Dra Carlos G. Malbrán, Buenos Aires, Argentina); Departamento de Microbiología y Parasitología,(UNAM, México D.F., Mexico); Centro de Biologia Molecular e Ambiental (CBMA), (Universidade do Minho, Braga, Portugal) and special thanks to Drs. Maria Lucia Taylor and Rocio Reyes-Montes members of the Mexican group (Departamento de Microbiología y Parasitología, UNAM), for their collaboration in this work. The work was supported by the MICOMOL-CYTED network and by Research Projects from Spanish Fondo de Investigaciones Sanitarias of the Instituto de Salud Carlos III (PI11/00412).

